- 腎臟病學(xué)(第4版)
- 王海燕 趙明輝主編
- 21212字
- 2025-03-06 17:32:59
第四章 炎癥與炎癥介質(zhì)在腎損傷中的作用
絕大多數(shù)腎臟疾病始發(fā)于或伴隨有免疫炎癥反應(yīng),免疫調(diào)控的紊亂是腎臟疾病發(fā)生發(fā)展一個重要的病理機制。當(dāng)腎臟組織遭受外界或自身原因?qū)е聯(lián)p傷后,血液來源的免疫細(xì)胞以及腎臟組織的固有細(xì)胞通過表達和釋放炎癥介質(zhì),介導(dǎo)炎癥反應(yīng)的發(fā)生[1]。炎癥反應(yīng)的強弱和持續(xù)時間長短在腎臟疾病的發(fā)生發(fā)展和轉(zhuǎn)歸過程中發(fā)揮關(guān)鍵作用。
在炎癥反應(yīng)過程中,起主導(dǎo)作用的是炎癥細(xì)胞和炎癥介質(zhì)。炎癥細(xì)胞激活后可合成和釋放大量的炎癥介質(zhì),如趨化因子、細(xì)胞因子等,一方面參與組織損傷或修復(fù);同時又募集和激活更多的炎癥細(xì)胞,進一步釋放炎癥介質(zhì)。炎癥細(xì)胞和炎癥介質(zhì)之間相互調(diào)節(jié),如果控制得當(dāng),則炎癥消退,疾病好轉(zhuǎn);如果炎癥持續(xù)存在或不斷放大,則加重組織損傷,疾病遷延不愈。
第一節(jié) 炎癥效應(yīng)細(xì)胞在腎臟病發(fā)生發(fā)展中的作用
廣義上講,所有參與炎癥反應(yīng)應(yīng)答的細(xì)胞均可稱為炎癥效應(yīng)細(xì)胞,其中包括來自于血液循環(huán)的免疫細(xì)胞,如中性粒細(xì)胞、單核/巨噬細(xì)胞、淋巴細(xì)胞、樹突狀細(xì)胞等;還有部分是來自于腎臟組織固有細(xì)胞,如內(nèi)皮細(xì)胞、系膜細(xì)胞、上皮細(xì)胞等(圖2-4-1-1)。近些年研究已明確證實,腎臟固有細(xì)胞不僅僅是炎癥損傷的被動受害者,同時也是主動參與者,其自身可表達或分泌多種炎癥介質(zhì)和細(xì)胞外基質(zhì),在炎癥反應(yīng)中發(fā)揮積極的作用[2]。
一、中性粒細(xì)胞
中性粒細(xì)胞是非特異性細(xì)胞免疫系統(tǒng)的主力軍,處于機體抵御病原微生物,特別是化膿性細(xì)菌入侵的第一線。當(dāng)炎癥發(fā)生時,中性粒細(xì)胞被趨化,從血液中游走出來聚集于炎癥部位,因其胞內(nèi)含有大量溶酶體酶,因此能將吞入的細(xì)菌和組織碎片分解,防止病原微生物在體內(nèi)擴散。當(dāng)中性粒細(xì)胞自身崩解時,釋放出多種溶酶體酶,溶解周圍組織而形成膿腫。中性粒細(xì)胞的細(xì)胞膜還能釋放出一種不飽和脂肪酸花生四烯酸,在酶的作用下,進一步生成一組旁分泌激素物質(zhì),如血栓素和前列腺素等,這類物質(zhì)對調(diào)節(jié)血管口徑和通透性有明顯的作用,還能引起炎癥反應(yīng)和疼痛,并影響血液凝固。中性粒細(xì)胞受細(xì)菌產(chǎn)物、抗原抗體復(fù)合物等作用時,細(xì)胞的顆粒內(nèi)容物向細(xì)胞外釋放。釋出的酸性蛋白酶和中性蛋白酶,可以分解血管基膜、腎小球基膜、結(jié)締組織的膠原蛋白與彈性蛋白以及血漿中的補體C5、C15和激肽原等。其分解產(chǎn)物有的又是中性粒細(xì)胞趨化因子,能吸引更多的中性粒細(xì)胞。此外,中性粒細(xì)胞還能分泌多種活性氧簇(ROS),活性氮簇(reactive nitrogen species,RNS)和趨化因子,吸引其他的炎癥細(xì)胞聚集,加重炎癥反應(yīng)。
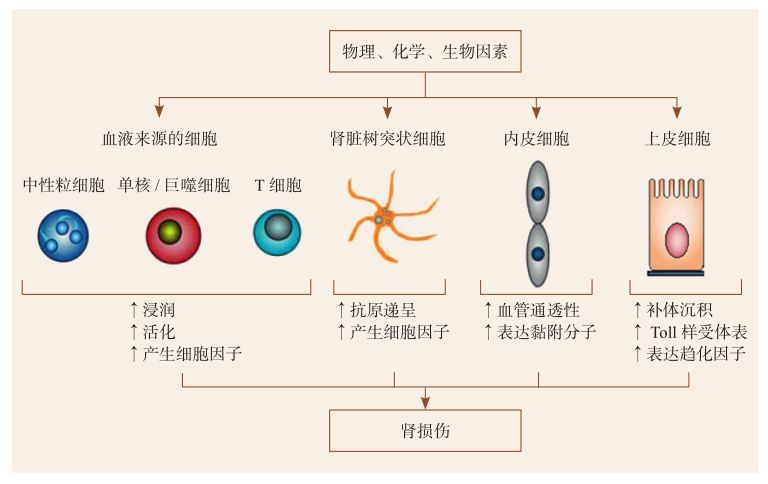
圖2-4-1-1 炎癥反應(yīng)介導(dǎo)腎臟組織損傷
在急性腎損傷的動物模型和病人腎臟組織活檢中均發(fā)現(xiàn)大量中性粒細(xì)胞的聚集。缺血-再灌注30分鐘后,中性粒細(xì)胞就大量聚集于腎臟皮髓交界處的腎小管周圍毛細(xì)血管網(wǎng)周圍。在一些黏附分子,如細(xì)胞間黏附分子-1(ICAM-1)、P-選擇素的作用下,中性粒細(xì)胞黏附于血管內(nèi)皮細(xì)胞,與紅細(xì)胞、血小板共同引起毛細(xì)血管腫脹、充血。中性粒細(xì)胞脫顆粒,釋放蛋白酶、過氧化物酶和細(xì)胞因子,同時產(chǎn)生活性氧,引起血管內(nèi)皮細(xì)胞和外髓質(zhì)區(qū)腎小管上皮細(xì)胞的損傷。浸潤到間質(zhì)的中性粒細(xì)胞通過增強血管通透性,損傷內(nèi)皮細(xì)胞、上皮細(xì)胞完整性而加重腎臟損傷[3](圖2-4-1-2)。有研究發(fā)現(xiàn),游走入間質(zhì)的中性粒細(xì)胞與聚集于血管內(nèi)壁的中性粒細(xì)胞有不同的細(xì)胞因子表達譜,如γ-干擾素(IFN-γ)、白細(xì)胞介素-4(IL-4)、白細(xì)胞介素-6(IL-6)、白細(xì)胞介素-10(IL-10)和腫瘤壞死因子-α(TNF-α)。此外,還有研究發(fā)現(xiàn),中性粒細(xì)胞浸潤程度與缺血-再灌注損傷嚴(yán)重程度呈一定相關(guān)性。
中性粒細(xì)胞還參與腎小球疾病的病理生理過程。當(dāng)系膜區(qū)或內(nèi)皮下有免疫復(fù)合物沉積時,常有中性粒細(xì)胞的聚集,中性粒細(xì)胞與免疫復(fù)合物接觸后可被激活,產(chǎn)生蛋白溶解酶,消化腎小球基底膜;中性粒細(xì)胞產(chǎn)生的ROS可誘導(dǎo)腎臟固有細(xì)胞表型發(fā)生轉(zhuǎn)變;中性粒細(xì)胞還可釋放髓系過氧化物酶,與腎小球毛細(xì)血管壁上的負(fù)電荷物質(zhì)結(jié)合,產(chǎn)生毒性更大的活性物質(zhì),損傷基底膜;此外,中性粒細(xì)胞還可生成并釋放前列腺素、白三烯等磷脂代謝產(chǎn)物,以及組胺、5-羥色胺等血管活性物質(zhì),最終造成腎小球病變,產(chǎn)生蛋白尿。
二、單核/巨噬細(xì)胞
正常情況下,在血液中分化發(fā)育成熟的單核細(xì)胞透過血管壁游走進入組織中,繼續(xù)增殖分化為組織巨噬細(xì)胞。巨噬細(xì)胞吞噬、消化體內(nèi)衰老、死亡的細(xì)胞和組織中的碎片;同時還分泌多種細(xì)胞因子,活化淋巴細(xì)胞,介導(dǎo)炎癥反應(yīng)[4]。
單核/巨噬細(xì)胞浸潤是腎小球和腎小管間質(zhì)疾病最具特征性的表現(xiàn)之一。無論從急性損傷到慢性進展性腎病,從炎性腎病(如腎小球腎炎、急性間質(zhì)性腎炎)到非炎性腎病(如糖尿病腎病、梗阻性腎病),從臨床到動物實驗,均可看到單核/巨噬細(xì)胞的浸潤,其浸潤程度通常被用來預(yù)測疾病的發(fā)展和轉(zhuǎn)歸[5]。
早在20世紀(jì)60年代,臨床研究就已發(fā)現(xiàn),在多種人類腎小球炎癥病變中存在大量巨噬細(xì)胞的浸潤。20世紀(jì)80年代發(fā)現(xiàn)單核/巨噬細(xì)胞是細(xì)胞新月體的組成部分。此外,在彌漫增殖性腎炎、抗腎小球基底膜(GBM)腎炎及狼瘡性腎炎的系膜區(qū),毛細(xì)血管腔內(nèi)均有明顯的單核/巨噬細(xì)胞浸潤。
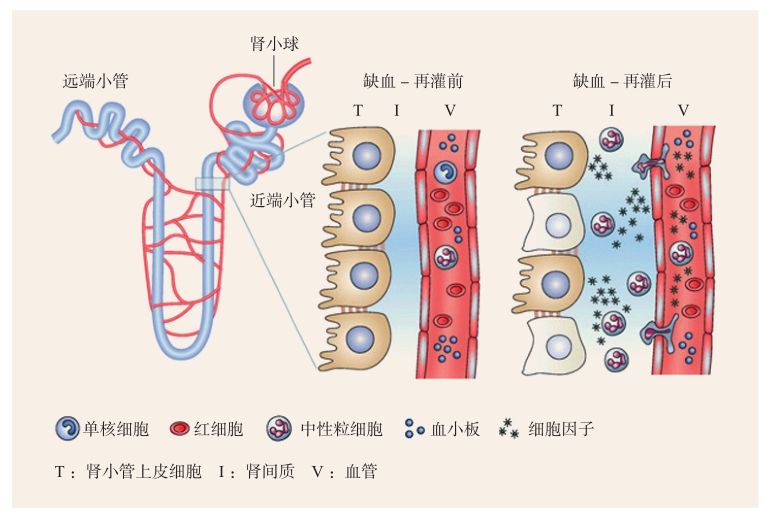
圖2-4-1-2 缺血-再灌后中性粒細(xì)胞介導(dǎo)腎小管上皮細(xì)胞損傷和炎癥反應(yīng)[3]
動物實驗提示,一些腎炎的腎小球細(xì)胞增殖和損傷與腎小球內(nèi)單核細(xì)胞的浸潤有關(guān)。預(yù)先除去單核細(xì)胞或應(yīng)用抗單核細(xì)胞血清可阻止或減輕蛋白尿的發(fā)生。有研究認(rèn)為單核細(xì)胞參與腎小球腎炎發(fā)病,與促進輔助性T細(xì)胞興奮,抑制性T細(xì)胞活性減弱有關(guān)。單核細(xì)胞在腎小球內(nèi)聚集后,釋放多種蛋白酶和膠原酶,破壞GBM;同時產(chǎn)生活性氧、花生四烯酸產(chǎn)物(如白三烯、血栓素A2),釋放內(nèi)皮素、血小板活化因子,直接損傷腎小球毛細(xì)血管;單核細(xì)胞產(chǎn)生細(xì)胞因子如IL-1、IL-6、TNF、轉(zhuǎn)化生長因子-β(TGF-β)、血小板源生長因子(PDGF)等刺激腎小球系膜細(xì)胞增殖;單核細(xì)胞還可作為抗原提呈細(xì)胞,參與腎小球局部炎癥反應(yīng)的啟動。
在糖尿病腎病早期,腎小球表達過量單核細(xì)胞趨化蛋白-1(MCP-1)、血管細(xì)胞黏附分子-1(VCAM-1)、ICAM-1等,吸引單核/巨噬細(xì)胞浸潤,浸潤的單核/巨噬細(xì)胞產(chǎn)生氧化應(yīng)激產(chǎn)物,活化金屬蛋白酶,促進一氧化氮(NO)合成,產(chǎn)生氧自由基,分泌細(xì)胞因子,損傷腎小球基底膜,加速腎小球硬化的進程[6,7]。在對糖尿病小鼠巨噬細(xì)胞浸潤與腎臟損害的關(guān)系研究中發(fā)現(xiàn),腎臟巨噬細(xì)胞浸潤與血糖水平明顯相關(guān)。提示持續(xù)高血糖可能是腎臟巨噬細(xì)胞積聚的重要推動因素[8]。
近年來對巨噬細(xì)胞更進一步的研究發(fā)現(xiàn),巨噬細(xì)胞其實是一群異質(zhì)性細(xì)胞,雖然它們均來自于血液中的單核細(xì)胞,但在組織不同的微環(huán)境作用下,分別向功能不同的M1型或M2型(又分為M2a、M2b、M2c)兩極極化,在炎癥反應(yīng)和組織重構(gòu)中發(fā)揮功能相反的作用[9]。
M1型即“經(jīng)典活化型”,主要指在有外來物感染或炎癥信號刺激下,經(jīng)過經(jīng)典的免疫途徑被誘導(dǎo)并活化,即在微生物產(chǎn)生的IFN-γ、脂多糖(LPS)、TNF-α等因子的作用下被活化,分泌IL-1β、IL-6、IL-12、IL-23、TNF-α等促炎因子,同時表達MHC-Ⅱ類分子,誘導(dǎo)Th1、Th17和CD4+T細(xì)胞活化,加劇炎癥反應(yīng)(圖2-4-1-3A)。
M2型是“選擇性活化型”,是在IL-4、IL-13等細(xì)胞因子作用下被誘導(dǎo)的,主要發(fā)揮抑制炎癥反應(yīng)的作用,避免M1型巨噬細(xì)胞造成的炎癥過度,使炎癥處于可控之中。M2型巨噬細(xì)胞的活化往往被認(rèn)為是繼發(fā)于初始免疫或次級免疫之后的。M2型巨噬細(xì)胞發(fā)揮炎癥抑制的作用主要依賴于分泌大量炎癥抑制因子,如IL-10、TGF-β等;同時還能夠分泌精氨酸酶1(arginase-1),可以通過抑制促炎因子NO的釋放來抑制炎癥反應(yīng);此外M2型巨噬細(xì)胞還能夠產(chǎn)生IL-1受體的拮抗劑,從而抑制IL-1、甘露糖受體的促炎作用(圖2-4-1-3A)。除了抑制炎癥反應(yīng)外,M2型巨噬細(xì)胞還有很強的吞噬作用和異物清除能力,能夠及時清除炎癥反應(yīng)中壞死的細(xì)胞和組織碎片;M2型巨噬細(xì)胞還能夠分泌營養(yǎng)因子,促進血管生成;產(chǎn)生纖連蛋白(FN)、基質(zhì)相關(guān)蛋白BIG-H3和胰島素樣生長因子-1(IGF-1),改善細(xì)胞外基質(zhì)(ECM)的重塑,促進創(chuàng)傷組織修復(fù),在組織重構(gòu)方面發(fā)揮重要作用[10,11]。近年來還發(fā)現(xiàn),脂肪組織中的M2型巨噬細(xì)胞還能夠分泌基質(zhì)金屬蛋白酶-9(MMP-9),可以減弱腎臟的纖維性病變[12]。
正常情況下,腎臟組織浸潤的M1、M2型巨噬細(xì)胞共同存在,各自發(fā)揮其生理功能,維持機體免疫穩(wěn)態(tài)。當(dāng)巨噬細(xì)胞M1、M2比例發(fā)生變化,其促炎、抗炎平衡被打破后,則導(dǎo)致腎臟組織損傷,疾病發(fā)生(圖2-4-1-3B)[13]。組織活檢發(fā)現(xiàn),在肥胖相關(guān)腎病的腎臟組織中,M1、M2的比例發(fā)生明顯變化,以M1型為主[14,15]。大量的M1型巨噬細(xì)胞分泌炎癥因子TNF-α,造成組織劇烈的炎癥反應(yīng)和損傷。TNF-α一方面通過激活NF-κB信號通路,降低腎臟細(xì)胞抗衰老蛋白Klotho的表達,引起腎臟細(xì)胞的損傷;同時還能夠促進脂肪細(xì)胞表達纖溶酶原激活劑抑制物(PAI-1),提高血漿中PAI-1濃度,從而減弱纖維蛋白降解,導(dǎo)致腎臟纖維化和終末期腎臟病[16]。TNF-α還能夠通過激活腎系膜細(xì)胞的p38MAPK信號通路,誘導(dǎo)MCP-1的表達[17]。MCP-1是一個重要募集單核細(xì)胞的趨化分子,使更多的單核細(xì)胞聚集于腎小球內(nèi)再分化為巨噬細(xì)胞;此外,M1型巨噬細(xì)胞釋放的TNF-α還能促進脂肪細(xì)胞釋放IL-6,進一步加重炎癥反應(yīng)和腎臟損傷[18]。
總的來說,在病變的急性期,受損的組織細(xì)胞分泌趨化因子,表達黏附分子,促使大量單核細(xì)胞從血液中游走出來而聚集于受損部位,此時的巨噬細(xì)胞以M1型為主,誘發(fā)炎癥反應(yīng),造成周圍組織損傷。為了控制炎癥反應(yīng),修復(fù)受損組織,M1型巨噬細(xì)胞逐漸轉(zhuǎn)為M2型。但若炎癥未能有效控制,轉(zhuǎn)為慢性期時,組織處于一種低水平炎癥狀態(tài),M2型持續(xù)存在并分泌大量TGF-β,則促進腎小球硬化、間質(zhì)纖維化,造成預(yù)后不良。事實上,巨噬細(xì)胞猶如一把“雙刃劍”,既能造成組織損傷,又能促進創(chuàng)傷修復(fù),如果能夠有效地對巨噬細(xì)胞的活性、表型加以調(diào)控和利用,有望成為治療腎臟病的新策略。
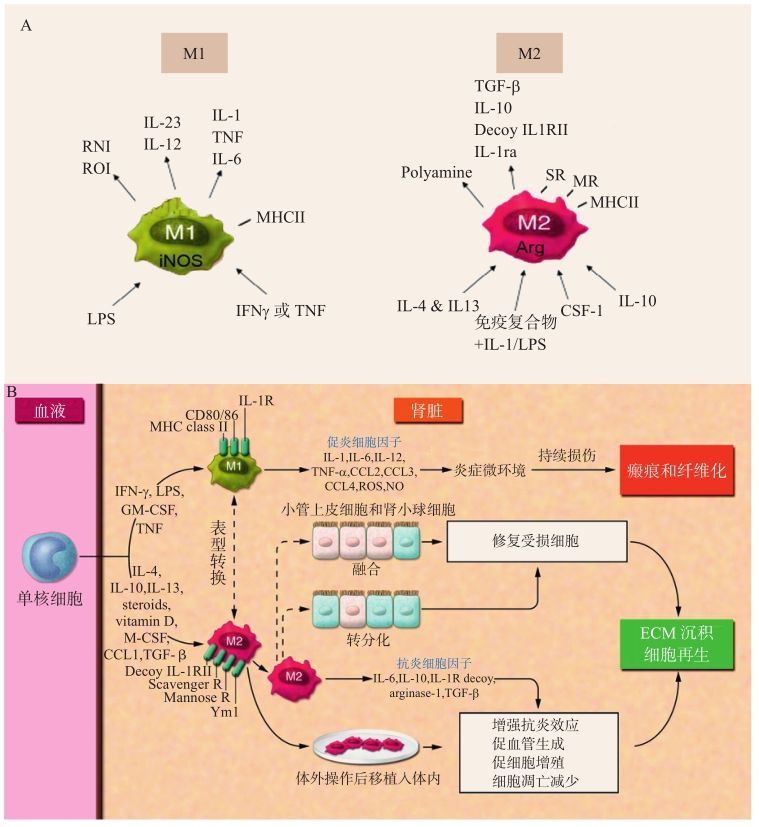
圖2-4-1-3 巨噬細(xì)胞的表型(A);腎臟受損后,巨噬細(xì)胞介導(dǎo)炎癥反應(yīng)和組織修復(fù)(B)[13]
三、T細(xì)胞
T細(xì)胞是淋巴細(xì)胞的主要組成,參與腎臟炎癥發(fā)生發(fā)展的細(xì)胞類型主要是CD8+及CD4+T細(xì)胞。這些細(xì)胞通過細(xì)胞黏附分子的介導(dǎo)在腎組織內(nèi)聚集和活化。它們可通過細(xì)胞毒作用直接殺傷細(xì)胞,或通過趨化或激活單核/巨噬細(xì)胞和自然殺傷(NK)細(xì)胞,誘導(dǎo)遲發(fā)型變態(tài)反應(yīng)造成腎臟損傷。此外,還可通過釋放各種細(xì)胞因子,參與及擴大炎癥反應(yīng)。CD8+T細(xì)胞能夠直接與靶細(xì)胞特異性結(jié)合,破壞靶細(xì)胞膜,直接殺傷靶細(xì)胞。
CD4+T細(xì)胞接受抗原刺激后首先分化為Th0細(xì)胞,Th0細(xì)胞在微環(huán)境的作用下,通過特異的轉(zhuǎn)錄因子調(diào)控,逐漸分化為Th1、Th2、Th17和Treg四個主要的亞群。各亞群分泌的細(xì)胞因子決定了每個亞群的免疫效應(yīng)功能,同時還調(diào)控各亞群的形成和擴增。Th1細(xì)胞主要通過分泌IFN、IL-2等細(xì)胞因子,介導(dǎo)細(xì)胞免疫,發(fā)生超敏反應(yīng);Th2細(xì)胞主要通過分泌IL-10、IL-4等細(xì)胞因子介導(dǎo)體液免疫(圖2-4-1-4)。生理條件下,機體的Th1細(xì)胞和Th2細(xì)胞處于相對平衡狀態(tài),在疾病狀態(tài)下,這種平衡被打破。臨床發(fā)現(xiàn),2型糖尿病患者存在Th1和Th2細(xì)胞亞群的失衡,在其腎臟病變發(fā)病的不同階段,存在不同的Th1和Th2優(yōu)劣勢。2型糖尿病并伴有大量白蛋白尿患者的Th1數(shù)和Th1/Th2比值低于正常對照組,而2型糖尿病伴微量白蛋白患者的Th1數(shù)和Th1/Th2比值高于正常對照組。在Ⅳ型狼瘡性腎炎患者中Th1/Th2比值是升高的,主要以Th1升高為主,這與Ⅳ型狼瘡性腎炎是以免疫復(fù)合物沉積介導(dǎo)的免疫反應(yīng)是一致的。臨床發(fā)現(xiàn)IgA腎病中有反復(fù)發(fā)作性肉眼血尿的患者Th1細(xì)胞比例及Th1/Th2值均較無此病史組的高。多數(shù)患者在肉眼血尿發(fā)生前伴有上呼吸道感染表現(xiàn),這可能與局部炎癥時Th1細(xì)胞免疫應(yīng)答增強有關(guān),同時由于炎癥的持續(xù)存在,刺激大量細(xì)胞因子的釋放(如IL-1、IL-6)作用于腎臟,進一步加重炎癥反應(yīng),導(dǎo)致肉眼血尿的發(fā)生。
Th17主要通過分泌IL-17A、IL-17F等參與機體炎癥和自身免疫反應(yīng)[19]。IgA腎病患者腎小管間質(zhì)IL-17的表達水平與畸形紅細(xì)胞計數(shù)、腎小管間質(zhì)病理呈正相關(guān),IL-17mRNA的表達與血肌酐呈正相關(guān)。以上結(jié)果提示IL-17可能是參與IgA腎病發(fā)病及進展的因素之一[20]。有研究表明,在IgA腎病患者中,IL-17可刺激單核細(xì)胞釋放IL-1β、TNF-α,而IL-1β則可以抑制IL-17的上述作用。而IL-1β、TNF-α是重要的致炎因子,提示IL-17可能是參與IgA腎病腎臟內(nèi)炎癥反應(yīng)的因素之一[21]。綜上所述,異常糖基化的IgA易于聚集,或與IgG聚集,沉積于腎臟系膜區(qū),從而活化系膜細(xì)胞,導(dǎo)致系膜細(xì)胞增生、釋放多種細(xì)胞因子。而這些細(xì)胞因子可以激活Th17細(xì)胞,使其釋放IL-17等,進一步介導(dǎo)腎臟的炎癥、免疫反應(yīng),引起腎臟損傷。對于異常糖基化的IgA與Th17細(xì)胞是否具有功能上的聯(lián)系,以及其確切的聯(lián)系機制,目前尚無研究。
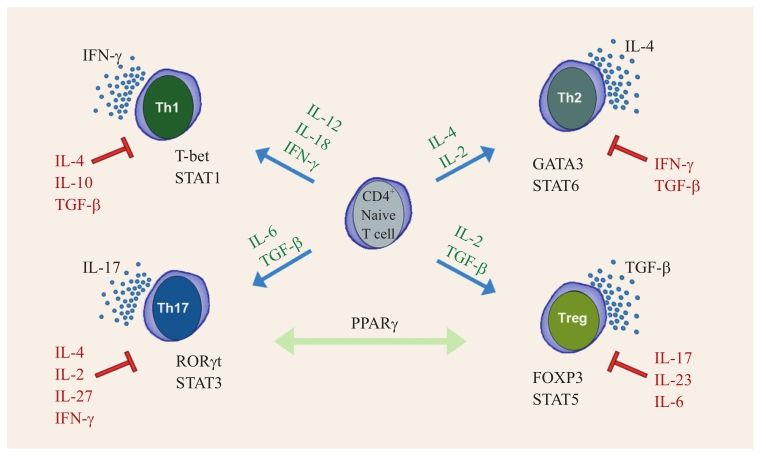
圖2-4-1-4 CD4+T細(xì)胞各亞群分化示意圖
調(diào)節(jié)性T淋巴細(xì)胞(regulatory T cell,Treg)則是指一類在分化過程中表面高表達CD25且具有免疫抑制功能的CD4+T淋巴細(xì)胞亞群。其抑制功能的發(fā)揮主要是通過分泌IL-10、TGF-β等細(xì)胞因子和細(xì)胞接觸機制來實現(xiàn)的[22]。在原發(fā)性腎病綜合征動物模型中,可以通過誘導(dǎo)Treg的產(chǎn)生,降低蛋白尿,緩解腎小球的病變,減少T淋巴細(xì)胞在腎臟的浸潤,這就表明Treg對腎臟有保護作用[23]。狼瘡性腎炎患者血液中,Treg細(xì)胞明顯降低,同時還伴有血清低表達TGF-β,尿液高表達TGF-β。經(jīng)激素治療后,Treg比例增加,提示Treg有可能成為狼瘡活動性的一個指標(biāo)[24]。目前2型糖尿病的發(fā)病機制并未明確,但國內(nèi)、外的研究結(jié)果均表明2型糖尿病與其血管并發(fā)癥的發(fā)生與免疫損傷相關(guān),在2型糖尿病腎病患者中Treg細(xì)胞較對照組顯著降低,而且隨著病情的加重,Treg的降低更加明顯,提示Treg細(xì)胞可能參與了2型糖尿病及其腎病的發(fā)生和發(fā)展,但其具體機制仍需進一步深入探討。
四、樹突狀細(xì)胞
樹突狀細(xì)胞(dendritic cells,DC)是機體中功能最強的專職抗原遞呈細(xì)胞,既能直接激活初始型T淋巴細(xì)胞(naive T cell),啟動早期特異性免疫應(yīng)答,又能誘導(dǎo)免疫耐受,已成為免疫學(xué)及相關(guān)領(lǐng)域的研究熱點[25]。根據(jù)來源,DC細(xì)胞可被分為兩類,即來源于髓系干細(xì)胞的髓樣樹突狀細(xì)胞(myeloid DC)和來源于淋巴系干細(xì)胞的淋巴樣樹突狀細(xì)胞(lymphoid DC)。這些DC因其組織分布情況或分化程度的不同而又有不同的名稱,位于腎臟組織中的DC稱為間質(zhì)樹突狀細(xì)胞(interstitial DC)。
有研究在IgA腎病、狼瘡性腎炎、急性腎損傷及單側(cè)輸尿管梗阻(UUO)模型中發(fā)現(xiàn)DC參與腎小管間質(zhì)炎癥反應(yīng),且DC向局部組織的遷徙與P-選擇素的黏附介導(dǎo)有關(guān)。糖尿病腎病患者外周血單核細(xì)胞向DC分化的能力顯著低于正常人;分化而成的DC能遞呈抗原,但亞群比例失調(diào)。說明糖尿病腎病患者樹突狀細(xì)胞誘導(dǎo)調(diào)節(jié)T細(xì)胞能力異常。在原發(fā)性腎病綜合征患者DC與T細(xì)胞相互作用時,DC激發(fā)T細(xì)胞活化的能力減弱,且傾向誘導(dǎo)Th向Th2分化,從而參與Th2占優(yōu)勢的內(nèi)環(huán)境形成。在終末期腎病患者中發(fā)現(xiàn),外周血單核細(xì)胞源性的DC發(fā)育不成熟,其趨化遷移功能以及外源性抗原遞呈能力下降,免疫激活能力降低,可能導(dǎo)致T細(xì)胞功能缺陷,從而最終誘導(dǎo)機體特異性的免疫耐受[26]。
五、腎臟固有細(xì)胞
1.內(nèi)皮細(xì)胞
內(nèi)皮細(xì)胞是位于循環(huán)血液與血管壁內(nèi)皮下組織間的單層細(xì)胞,是維持毛細(xì)血管結(jié)構(gòu)完整性并進行血液和組織間物質(zhì)交換的選擇性通透屏障。內(nèi)皮細(xì)胞可感知血液中的炎性刺激、激素水平和壓力等信號的變化,并通過分泌多種血管活性物質(zhì)對這些信號作出反應(yīng)。
當(dāng)內(nèi)皮細(xì)胞受損后,其抗凝活性下降,促進血小板、中性粒細(xì)胞的黏附與聚集,毛細(xì)血管微血栓形成;同時,內(nèi)皮細(xì)胞表達多種黏附分子,如血小板/內(nèi)皮細(xì)胞黏附分子-1(platelet-endothelial cell adhesion molecule-1,PECAM-1)、血管內(nèi)皮鈣黏蛋白(vasoendothelial-cadherin,VE-Cadherin)和CD99等,介導(dǎo)白細(xì)胞從血管內(nèi)遷移至炎癥損傷部位,經(jīng)過一系列胞質(zhì)蛋白酪氨酸磷酸化過程而致活化。目前已有實驗證明,應(yīng)用PECAM-1、CD99的抗體阻斷它們的作用,可明顯減少炎癥細(xì)胞的外滲;但應(yīng)用VE-Cadherin的抗體,則發(fā)現(xiàn)炎癥細(xì)胞外滲增加[27]。在內(nèi)膜新生血管中,VECadherin的表達下調(diào)與內(nèi)膜炎癥密切相關(guān)。此外,內(nèi)皮細(xì)胞還有合成多種炎癥因子的潛能,損傷或激活的內(nèi)皮細(xì)胞還產(chǎn)生促炎因子、急性期蛋白、C-反應(yīng)蛋白和氧化低密度脂蛋白等,介導(dǎo)局部炎癥的發(fā)生;內(nèi)皮細(xì)胞同時上調(diào)表達選擇素、趨化因子等多種細(xì)胞因子,進一步吸引組織中的炎癥細(xì)胞聚集于腎小球,共同發(fā)揮炎癥效應(yīng)。總之,內(nèi)皮細(xì)胞特性的改變,導(dǎo)致凝血纖溶的失衡、炎癥反應(yīng)的發(fā)生,是加重腎臟疾病進程,促進腎臟組織纖維化的重要原因[28]。
對糖尿病腎病的發(fā)病機制研究發(fā)現(xiàn),內(nèi)皮功能障礙是其發(fā)病的重要環(huán)節(jié)之一。血糖持續(xù)異常升高導(dǎo)致細(xì)胞組織損傷,血管內(nèi)皮細(xì)胞是首要受害者。受損的內(nèi)皮細(xì)胞表達細(xì)胞間黏附因子和血管黏附因子,分泌炎癥因子,對內(nèi)皮細(xì)胞的活化、白細(xì)胞黏附以及在免疫性小血管炎的發(fā)生中有重要作用,參與炎癥反應(yīng)、促進血栓形成[29]。糖尿病患者血液中可檢測到內(nèi)皮細(xì)胞抗體(anti-endothelial cell antibody,AECA),且檢出率隨著病程的發(fā)展而增加。該抗體對應(yīng)的抗原是位于血管內(nèi)皮細(xì)胞表面的異質(zhì)性抗原,在腎小管周圍毛細(xì)血管、腎小球毛細(xì)血管均有表達。AECA可介導(dǎo)免疫損傷進一步加重內(nèi)皮細(xì)胞功能障礙,促進糖尿病腎病發(fā)生發(fā)展[30]。
2.系膜細(xì)胞
系膜細(xì)胞具有收縮、吞噬、產(chǎn)生細(xì)胞外基質(zhì)等多種功能,在維持腎臟正常生理功能及腎臟病變的發(fā)生發(fā)展中具有重要作用。生理條件下,系膜細(xì)胞增殖緩慢,分泌少量細(xì)胞外基質(zhì),且細(xì)胞外基質(zhì)的分泌與降解維持動態(tài)平衡。在炎癥和損傷等病理條件下,系膜細(xì)胞可被激活,表現(xiàn)為過度增殖同時大量分泌細(xì)胞外基質(zhì)和炎癥介質(zhì)[31]。
系膜增生性腎炎患者的腎小球系膜區(qū)有免疫球蛋白和補體C3的沉積引起系膜細(xì)胞增殖。當(dāng)系膜組織清除能力下降時,單核吞噬細(xì)胞系統(tǒng)功能受損,免疫復(fù)合物滯留于系膜區(qū)不能及時被清除,引起炎癥反應(yīng)。系膜細(xì)胞是炎癥介質(zhì)作用的靶細(xì)胞,通過自分泌和旁分泌方式,激活淋巴細(xì)胞,異常分泌細(xì)胞因子,促進系膜細(xì)胞增生;同時,系膜細(xì)胞還產(chǎn)生大量細(xì)胞外基質(zhì),細(xì)胞外基質(zhì)又通過細(xì)胞表面的受體和整合素信號通路影響細(xì)胞功能[32]。
系膜細(xì)胞活化是腎小球?qū)Χ喾N有害刺激反應(yīng)的共同結(jié)果,免疫復(fù)合物、激活的補體成分、脂多糖、細(xì)胞因子、血管活性肽等均可引起系膜細(xì)胞活化[33]。以炎癥為主的腎小球疾病,如IgA腎病、狼瘡性腎炎、糖尿病腎病等,在早期常伴有腎小球系膜增生,系膜細(xì)胞的異常增殖并繼發(fā)TNF-α、IL-6、MCP-1等炎癥介質(zhì)的釋放,細(xì)胞外基質(zhì)FN沉積,上述病理過程是腎小球硬化、使腎小球疾病走向終末期的中心環(huán)節(jié)之一[34]。在實驗動物腎病模型中,也常常發(fā)現(xiàn)系膜增生并伴隨細(xì)胞外基質(zhì)增多和炎癥介質(zhì)分泌的病理學(xué)特征。
3.腎小管上皮細(xì)胞
腎小管上皮細(xì)胞在腎臟功能中有著重要作用,然而它也是許多先天性疾病、代謝性疾病和炎癥反應(yīng)的主要損傷部位。正常的腎小管上皮細(xì)胞具有旺盛的代謝活性和潛在的增殖能力,并分泌多種細(xì)胞因子。在疾病狀態(tài)下,腎小管上皮細(xì)胞極易發(fā)生結(jié)構(gòu)和功能損傷,釋放炎癥介質(zhì),參與間質(zhì)炎癥、纖維化等過程。
腎組織急性缺血、缺氧,再恢復(fù)血液灌注時,腎小管上皮細(xì)胞受損,分泌一系列促炎因子和促纖維化因子,引起間質(zhì)炎癥反應(yīng),導(dǎo)致腎小管上皮細(xì)胞凋亡,腎小管萎縮、腎間質(zhì)纖維化。在早期急性腎小管壞死期,有大量中性粒細(xì)胞浸潤,產(chǎn)生ROS、表達黏附分子、分泌炎性趨化因子等。此時腎小管上皮Toll樣受體-2(TLR2)表達增強,敲除TLR2基因,可以避免缺血導(dǎo)致的腎功能異常、中性粒細(xì)胞聚集、小管上皮細(xì)胞凋亡以及MCP-1、TNF-α、IL-6和IL-1的產(chǎn)生增加。在急性腎衰竭恢復(fù)期,以巨噬細(xì)胞和T細(xì)胞浸潤為主。炎癥細(xì)胞的大量浸潤與小管上皮細(xì)胞黏附分子表達增加有關(guān),如選擇素、整合素等。動物體內(nèi)研究發(fā)現(xiàn),應(yīng)用ICAM-1抗體中和或敲除ICAM-1基因?qū)毙匀毖鸬哪I損傷有一定保護作用。
在慢性炎癥過程中,長期的炎性刺激使小管上皮細(xì)胞受損,產(chǎn)生一系列趨化因子,募集炎癥細(xì)胞于小管周圍,造成局部的炎癥微環(huán)境。這種微環(huán)境中的纖維化因子、細(xì)胞因子促使上皮細(xì)胞去分化,轉(zhuǎn)化為間充質(zhì)細(xì)胞,活化肌成纖維細(xì)胞,分泌大量膠原等細(xì)胞外基質(zhì)沉積于小管周圍,壓迫、擠壓小管并破壞組織結(jié)構(gòu)。炎性浸潤和上皮-間充質(zhì)轉(zhuǎn)分化(EMT)形成一個惡性循環(huán),加重小管周圍炎癥,小管細(xì)胞去分化和間質(zhì)纖維化[35,36]。在這個惡性循環(huán)中,分化抑制物1(inhibitor of differentiation 1,Id1)分子發(fā)揮著重要作用。腎小管上皮細(xì)胞受損時,近端小管上皮細(xì)胞Id1表達迅速增加,激活上皮細(xì)胞中NF-κB信號通路,并促進趨化因子RANTES表達,促進炎癥細(xì)胞聚集。同時Id1還上調(diào)轉(zhuǎn)錄因子Snail1的表達,促進小管上皮細(xì)胞去分化。在Id1敲除的小鼠UUO模型中,可明顯觀察到小管周圍炎癥減輕,小管分泌RANTES減少;同時肌成纖維細(xì)胞活化減弱,細(xì)胞外基質(zhì)沉積減少[37]。
4.足細(xì)胞
足細(xì)胞是腎小球中一種高度終末分化細(xì)胞,附著于腎小球基底膜的外側(cè),連同腎小球基底膜和毛細(xì)血管內(nèi)皮一起構(gòu)成了腎小球血液濾過屏障,在維持腎小球通透性上起著關(guān)鍵作用。足細(xì)胞易受到多種因素造成的損傷,如免疫、炎癥、毒物、感染、代謝、環(huán)境、遺傳等因素。足細(xì)胞損傷是腎小球損傷的中心環(huán)節(jié),直接導(dǎo)致蛋白尿形成,腎小球硬化、纖維化。
多種炎癥物質(zhì)可以刺激足細(xì)胞,如TGF-α、IL-1、白三烯等可以促進足細(xì)胞增殖,TGF-β、IL-2、PGE2、脂多糖等則抑制足細(xì)胞增殖。在足細(xì)胞受到刺激后又能分泌補體、趨化因子、共刺激分子等多種炎癥介質(zhì),同時還表達細(xì)胞因子受體、Toll樣受體等,對炎癥反應(yīng)作出應(yīng)答。
NF-κB介導(dǎo)的炎癥信號通路的活化在足細(xì)胞損傷中也發(fā)揮著重要作用。足細(xì)胞中該通路的失活對小鼠的正常發(fā)育無任何影響,但在通過腹腔注射羊抗兔腎小球抗體誘導(dǎo)腎毒性腎炎模型中發(fā)現(xiàn),該通路的失活明顯可以減少腎小球內(nèi)單核/巨噬細(xì)胞、中性粒細(xì)胞、T細(xì)胞的浸潤,降低趨化因子MCP-1/CCL-2、CCL-7的分泌,促進受損足細(xì)胞形態(tài)的恢復(fù),減少蛋白尿產(chǎn)生[38]。
病毒感染引起免疫復(fù)合物沉積是導(dǎo)致腎小球腎炎的常見原因之一,腎小球系膜細(xì)胞、腎小球內(nèi)皮細(xì)胞識別病毒核酸物質(zhì),釋放IFN-α、IFN-β,抑制腎臟前體細(xì)胞分化為成熟的足細(xì)胞;同時IFN-β還能增強足細(xì)胞通透性,引起足細(xì)胞死亡。在多柔比星誘導(dǎo)的腎病小鼠模型中,注射IFN-α、IFN-β后小鼠蛋白尿加重、腎小球區(qū)域巨噬細(xì)胞浸潤增多,腎小球硬化明顯。進一步研究發(fā)現(xiàn),IFN-β可誘導(dǎo)足細(xì)胞進行有絲分裂,但該過程并非是促進足細(xì)胞增殖、細(xì)胞數(shù)目增多,而是促使足細(xì)胞從基底膜上脫落、死亡,導(dǎo)致足細(xì)胞數(shù)目減少,腎小球基底膜區(qū)域性裸露,裂隙隔膜遭到破壞,大量蛋白從此濾過,使腎小球形成“高濾過、高灌注和高跨膜壓”,最終形成腎小球硬化、腎功能進行性喪失[39]。
糖尿病腎病是一種由代謝紊亂引起的炎癥性疾病,既往研究認(rèn)為腎小球基底膜成分改變及細(xì)胞外基質(zhì)聚積是糖尿病腎病的關(guān)鍵性改變。近年來研究發(fā)現(xiàn),足細(xì)胞超微結(jié)構(gòu)改變及其相關(guān)分子表達變化在糖尿病腎病蛋白尿產(chǎn)生及發(fā)展過程中發(fā)揮重要作用。在高血糖、氧化應(yīng)激、腎小球血流動力學(xué)改變等多種因素刺激下,MCP-1表達明顯上調(diào)。MCP-1通過調(diào)控TGF-β和NF-κB信號通路促進腎小球纖維化,導(dǎo)致腎臟病變。TGF-β和Smad7可促進足細(xì)胞發(fā)生凋亡,兩者相互具有協(xié)同效應(yīng),TGF-β1受體可通過p38MAPK和caspase3介導(dǎo)細(xì)胞凋亡,使細(xì)胞外基質(zhì)降解減少、合成增多;而Smad7可通過調(diào)控NF-κB活性來介導(dǎo)足細(xì)胞凋亡[40]。基質(zhì)金屬蛋白酶是鈣、鋅依賴的內(nèi)肽酶家族成員,在生理和病理狀態(tài)下的組織重建過程中發(fā)揮作用。在高糖環(huán)境下,足細(xì)胞MMP-9活性降低,使系膜基質(zhì)降解減少,在一定程度上導(dǎo)致腎小球毛細(xì)血管基底膜增厚和系膜基質(zhì)增多[41]。
第二節(jié) 炎癥介質(zhì)在腎臟損害中的作用
免疫反應(yīng)激活炎癥細(xì)胞,使之表達或釋放多種炎癥介質(zhì),介導(dǎo)腎臟組織損傷;炎癥介質(zhì)吸引和募集更多的炎癥細(xì)胞聚集,進一步擴大炎癥反應(yīng),加重組織損傷。在這一系列過程中,涉及的炎癥介質(zhì)種類繁多,作用復(fù)雜。
一、趨化因子
趨化因子(chemokine)是一類一級結(jié)構(gòu)相似,主要對白細(xì)胞具有化學(xué)趨化作用等多種生物學(xué)效應(yīng)的小分子蛋白,在機體的防御和炎癥反應(yīng)等方面起著重要的調(diào)節(jié)作用。根據(jù)其N末端半胱氨酸殘基的相對位置,主要分為CC、CXC、XC、CX四個亞家族(表2-4-2-1)。趨化因子介導(dǎo)白細(xì)胞的遷移是通過作用于靶細(xì)胞上相應(yīng)的受體來完成的,目前已明確有20多種趨化因子受體(表2-4-2-1),均屬于七次跨膜轉(zhuǎn)運G蛋白偶聯(lián)受體超家族[42]。
不同的趨化因子及其相應(yīng)受體表達于不同類型的細(xì)胞(圖2-4-2-1),總的來說,單核細(xì)胞表達受體CCR1、CCR2、CCR5,因此受到CCL的趨化;而粒細(xì)胞因表達CXCR1、CXCR2,所以受到CXCL的作用而被募集。Th1和NK細(xì)胞則通過膜表面受體CXCR3,CXCR6,CCR5和CX3CR1與趨化因子結(jié)合而參與Ⅰ型細(xì)胞因子(IL-2、TNF-γ)介導(dǎo)的炎癥反應(yīng)。Th2細(xì)胞則通過CCR3、CCR4和CCR8參與Ⅱ型細(xì)胞因子(IL-4、IL-5和IL-13)介導(dǎo)的炎癥反應(yīng)。CCR6和CXCR3則主要表達于Th17細(xì)胞上,CCR4–8、CXCR3、CXCR6則主要在Tregs上。在腎臟組織中,內(nèi)皮細(xì)胞、足細(xì)胞、系膜細(xì)胞、小管上皮細(xì)胞、間質(zhì)成纖維細(xì)胞在受到一定刺激后均可以產(chǎn)生趨化因子。
缺血-再灌注損傷時,中性粒細(xì)胞、單核細(xì)胞/巨噬細(xì)胞迅速聚集于腎臟損傷部位;此過程有賴于一系列特定趨化因子的表達,如中性粒細(xì)胞的聚集依賴于CXC亞家族的IL-8/CXCL8,Gro-α/CXCL1,Gro-β/CXCL2的高表達;而單核/巨噬細(xì)胞則依賴于CC亞家族的MCP-1/CCL-2和CX亞家族fractalkine/CX3CL1的表達。隨后,CC亞家族的RANTES/CCL5表達也逐漸升高,募集淋巴細(xì)胞和更多的單核/巨噬細(xì)胞聚集(圖2-4-2-1)。近年來也有報道,缺血-再灌注損傷時,MCP-1/CCL-2通過受體CCR2可以募集Treg細(xì)胞聚集于受損部位,并發(fā)揮一定的保護作用。DC細(xì)胞是連接初始免疫反應(yīng)和繼發(fā)免疫反應(yīng)的一個重要橋梁,腎臟組織發(fā)生缺血-再灌注損傷后,DC細(xì)胞產(chǎn)生促炎因子TNF-α、IL-6、CCL及趨化因子MCP-1/CCL2和RANTES/CCL5,這些炎癥介質(zhì)進一步夠募集更多的單核/巨噬細(xì)胞于損傷部位。受損的內(nèi)皮細(xì)胞分泌fractalkine/CX3CL1,可增強巨噬細(xì)胞的浸潤[42]。
臨床和動物模型已有大量證據(jù)表明,在慢性腎損傷的進展過程中,趨化因子及其相應(yīng)受體在募集炎癥細(xì)胞過程中發(fā)揮重要作用[43]。臨床發(fā)現(xiàn),很多腎小球腎炎患者尿液中IL-8/CXCL8水平明顯高于正常人[44];在膜增生性腎炎、狼瘡性腎炎、新月體性腎小球腎炎患者的腎小球和腎間質(zhì)處有大量CXCR1陽性的中性粒細(xì)胞聚集。動物實驗發(fā)現(xiàn),采用抗IL-8/CXCL8的抗體可以減少炎癥細(xì)胞的聚集和蛋白尿的產(chǎn)生。在慢性腎臟損傷中,除了有大量中性粒細(xì)胞、單核/巨噬細(xì)胞和樹突狀細(xì)胞浸潤外,還有大量T細(xì)胞聚集。新月體腎小球腎炎患者尿液中MIP-1/CCL3顯著升高,組織活檢也發(fā)現(xiàn),表達其相應(yīng)受體CCR1和CCR5的CD3+T細(xì)胞大量聚集于腎小球和腎間質(zhì)部位[45,46]。
表2-4-2-1 趨化因子的分類及其相應(yīng)的受體[42]
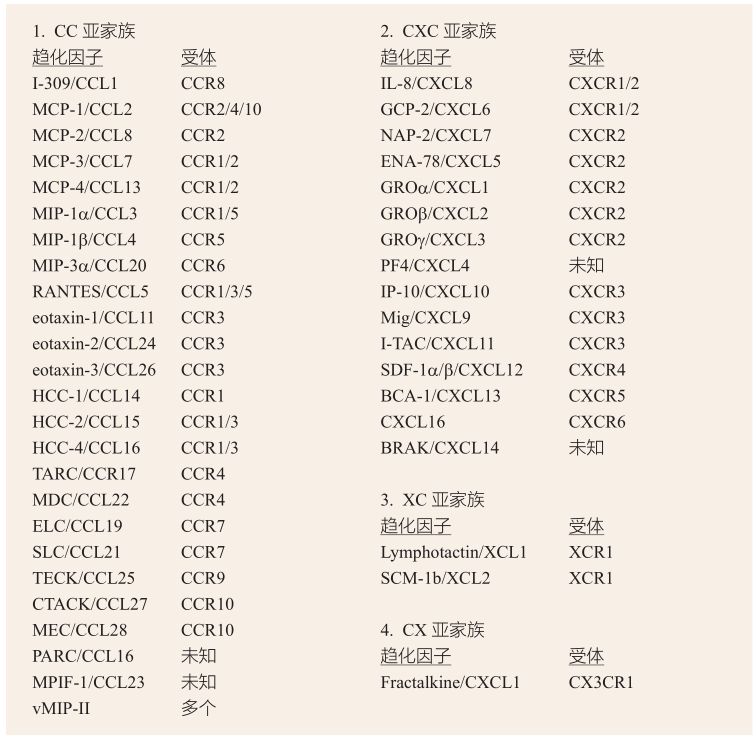
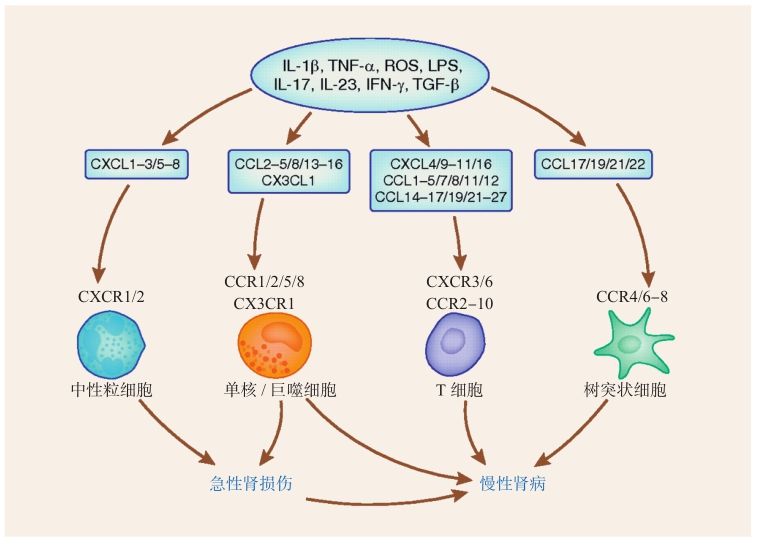
圖2-4-2-1 炎癥過程中趨化因子通過受體募集炎癥細(xì)胞,介導(dǎo)急、慢性腎損傷[42]
二、細(xì)胞因子
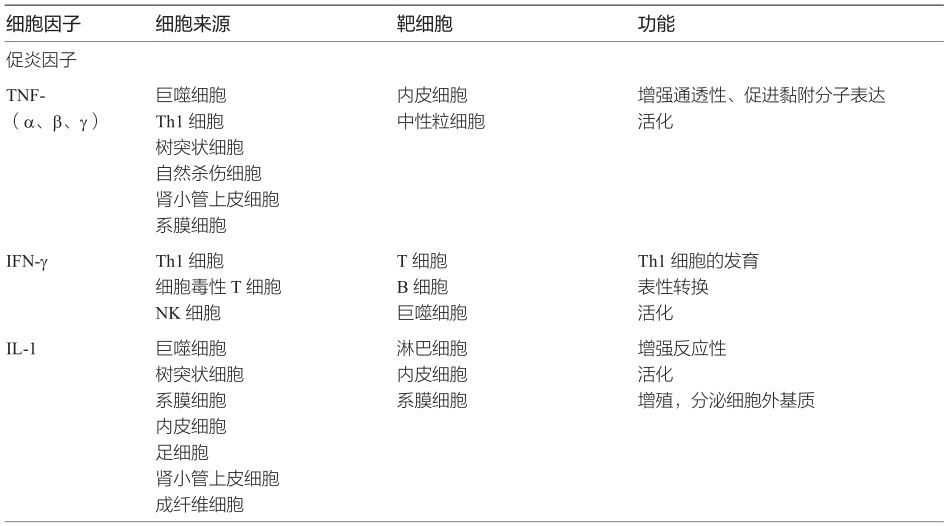
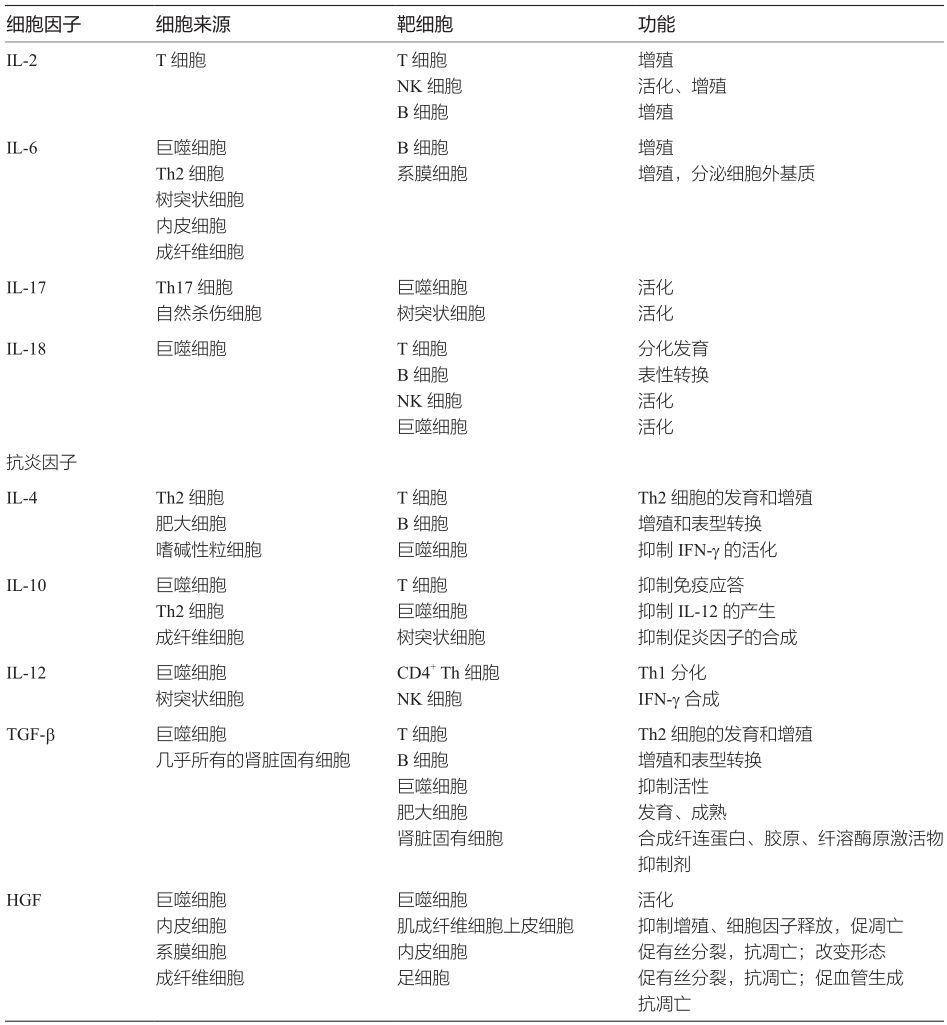
細(xì)胞因子是指由活化的炎癥細(xì)胞和某些基質(zhì)細(xì)胞分泌的、介導(dǎo)和調(diào)節(jié)免疫、炎癥反應(yīng)的多肽,是除免疫球蛋白和補體外的非特異性免疫效應(yīng)物質(zhì)(表2-4-2-2)。正常腎組織少量表達某些細(xì)胞因子,如TGF-β1,其生理意義目前還不清楚,推測可能與維持正常腎臟結(jié)構(gòu)和功能有關(guān)。當(dāng)腎臟疾病發(fā)生時,炎癥細(xì)胞和腎臟固有細(xì)胞產(chǎn)生多種細(xì)胞因子,根據(jù)功能主要分為兩類:一類是促炎因子,以Th1細(xì)胞表達為主,啟動或促進腎臟組織炎癥反應(yīng),加重組織損傷;另一類是抗炎因子,主要由Th2和Treg細(xì)胞表達,參與腎臟的自身防御,促進炎癥消散和組織修復(fù)。這對“攻擊”和“防御”因子的對決,決定著腎臟疾病的轉(zhuǎn)歸。
表2-4-2-2 細(xì)胞因子分類及功能
續(xù)表
1.促炎細(xì)胞因子
促炎因子是一系列可以促進炎癥反應(yīng)的細(xì)胞因子的總稱,包括有TNF、IFN-γ、IL-1,IL-2,IL-6,IL-17,IL-18等,可由多種細(xì)胞產(chǎn)生,特別是Th1細(xì)胞、巨噬細(xì)胞。這類細(xì)胞因子促進NO和一些炎性介質(zhì)的合成與釋放,活化細(xì)胞毒性T細(xì)胞、NK細(xì)胞和巨噬細(xì)胞,發(fā)揮促進炎癥反應(yīng)的作用[47]。
TNF主要由單核/巨噬細(xì)胞、淋巴細(xì)胞產(chǎn)生,不僅對腫瘤細(xì)胞有殺傷作用,還是機體炎癥反應(yīng)的重要因子。根據(jù)其來源和功能不同,可分為TNF-α、TNF-β、TNF-γ三類。在順鉑或內(nèi)毒素誘導(dǎo)的急性腎損傷模型中,TNF-α是介導(dǎo)組織損傷的重要因素,抑制其表達能夠減輕腎毒性作用[48,49]。在腎毒血清所致的新月體腎炎模型中,抗TNF-α治療可以明顯降低蛋白尿、減少尿液MCP-1含量,使血清肌酐和腎小球、腎小管間質(zhì)的瘢痕減少。在腎毒性腎病小鼠模型中,給小鼠體內(nèi)注射IL-1及TNF,促進腎臟損傷;注射IL-1受體拮抗物或可溶性TNF受體以阻斷IL-1或TNF-α,可以減輕腎組織病理改變,減少蛋白尿產(chǎn)生,保護腎功能[50]。在狼瘡腎炎、抗中性粒細(xì)胞胞質(zhì)抗體相關(guān)性腎炎、抗腎小球基膜性腎炎等疾病中,浸潤的白細(xì)胞釋放的TNF-α、IL-1β可引起腎臟固有細(xì)胞的增殖,刺激其表達黏附分子并生成過多的細(xì)胞外基質(zhì)和其他炎癥介質(zhì)。另有報道,腎小管間質(zhì)性腎炎、抗GBM腎炎、免疫復(fù)合物腎炎的發(fā)病機制均與IL-1有關(guān)[51,52]。
IL-6除了可以加重炎癥反應(yīng)外,還可以促進系膜細(xì)胞增殖和系膜基質(zhì)增多,采用抗IL-6單克隆抗體技術(shù)的研究發(fā)現(xiàn),系膜增生性腎小球腎炎的腎小球IL-6分泌增多,且IL-6的增加與增殖的系膜細(xì)胞數(shù)有關(guān)。非腎病或輕微腎病患者的尿中檢測不到IL-6,約7.4%的膜性腎病患者尿中IL-6為陽性,而系膜增生性腎小球腎炎則有50%的患者尿中可以檢測到IL-6,且尿中IL-6的高低與該病的嚴(yán)重程度成正相關(guān)。Dohi等隨訪了IgA腎病患者尿IL-6活性、臨床指標(biāo)和組織病理學(xué)改變,結(jié)果顯示,尿中IL-6活性持續(xù)增高的患者,IgA腎病的組織病理學(xué)改變有惡化趨勢;而隨訪10個月期間尿中未檢測到IL-6的患者,其組織學(xué)變化有所改善。尿中IL-6活性的檢測不僅可用于原發(fā)性腎小球疾病的鑒別診斷,而且可以作為反映IgA腎病進展的輔助手段[53]。
IL-17、IL-18與多種免疫性腎炎密切相關(guān)。IL-17在狼瘡腎炎小鼠血清和腎臟組織中的表達顯著增加,應(yīng)用抗IL-17抗體可抑制狼瘡腎炎的炎癥反應(yīng),減輕組織病理損害。臨床發(fā)現(xiàn)新月體腎炎和狼瘡腎炎患者的血、尿IL-18水平明顯增高,且與疾病的活動程度呈正相關(guān)[54]。此外,小鼠體內(nèi)研究還發(fā)現(xiàn),腎小管急性壞死時尿液中有大量IL-18,由于該結(jié)果穩(wěn)定可靠,檢測方法簡單,因此可作為急性腎小管壞死近端小管損傷的標(biāo)志。
2.抗炎細(xì)胞因子
與促炎癥細(xì)胞因子相比,目前對抗炎細(xì)胞因子的認(rèn)識還很有限。如同炎癥的啟動一樣,炎癥的消散也是一種動態(tài)的、有控制的、涉及多種抗炎癥介質(zhì)的過程。腎臟炎癥被啟動后,白細(xì)胞和血小板釋放致病介質(zhì)活化腎臟固有細(xì)胞;而腎臟固有細(xì)胞一旦被活化,又可以產(chǎn)生一系列“自我滅活”或“自我抑制”因子。這種自我調(diào)節(jié)方式構(gòu)成了控制腎臟炎癥過程的防御系統(tǒng),其中抗炎細(xì)胞因子是一個重要的組成部分。抗炎細(xì)胞因子同樣可由多種細(xì)胞產(chǎn)生,以Th2細(xì)胞為主,目前較為明確的有IL-4,IL-10,IL-12,TGF-β,肝細(xì)胞生長因子(HGF)等[55]。
體內(nèi)、體外研究發(fā)現(xiàn),抗炎細(xì)胞因子通過多種方式抑制炎癥反應(yīng),有的抑制促炎癥介質(zhì)的生成;有的刺激促炎癥介質(zhì)拮抗物的生成;有的降低腎細(xì)胞對促炎癥介質(zhì)的反應(yīng);有的抑制腎細(xì)胞的增殖。例如IL-4可以抑制巨噬細(xì)胞的活化,減少IFN-γ的釋放;IL-10可以抑制IL-1誘導(dǎo)的系膜細(xì)胞增生和ICAM-1的表達,同時還抑制脂多糖刺激的系膜細(xì)胞IL-1與TNF-α的產(chǎn)生。在新月體腎小球腎炎和系膜增生性腎炎模型中發(fā)現(xiàn),聯(lián)合使用重組鼠IL-4、IL-10可減少腎小球纖維蛋白的沉積和巨噬細(xì)胞、T細(xì)胞的浸潤,顯著降低新月體的形成,抑制系膜細(xì)胞增生,減少蛋白尿的產(chǎn)生;同時血清IgG2、IgG3同型免疫球蛋白降低,提示IL-4、IL-10通過選擇性抑制Th1免疫應(yīng)答延緩腎小球腎炎的發(fā)展。新月體腎小球腎炎模型中,IL-4或IL-10基因缺陷大鼠的病情較野生型嚴(yán)重[53]。TGF-β可以通過調(diào)控不同的下游分子來發(fā)揮不同的生物學(xué)功能;例如通過調(diào)控Smad2、Smad3和microRNA分子增強細(xì)胞外基質(zhì)分泌,促進EMT和腎纖維化的發(fā)生;同時還通過Smad4和Smad7調(diào)控NF-κB信號通路而抑制腎臟組織炎癥反應(yīng)[56]。HGF可以作用于腎臟組織中多種細(xì)胞,例如在慢性腎衰的早期階段,可以作用于肌成纖維細(xì)胞和腎小管上皮細(xì)胞,抑制促炎因子、促纖維化因子的釋放[57]。
通過藥物干預(yù)或基因治療以重建腎臟的自身防御體系,將是腎臟病治療的一個新途徑。已有研究證實,小鼠體內(nèi)注射IL-4或在腎小球內(nèi)轉(zhuǎn)移表達IL-10的系膜細(xì)胞載體可以減輕抗基底膜腎炎的蛋白尿和腎組織損害[58]。對早期新月體腎小球腎炎小鼠使用IL-4治療,可減少蛋白尿的產(chǎn)生和腎小球炎癥損傷;即使在炎癥反應(yīng)建立后應(yīng)用,仍能有效減輕腎臟損害。
然而應(yīng)該看到,對腎臟抗炎癥因子網(wǎng)絡(luò)的熟悉還很有限,抗炎細(xì)胞因子分子間的相互關(guān)系以及它們在病理狀態(tài)下的表達調(diào)控仍有待闡明。某些抗炎細(xì)胞因子具有“雙刃劍”的作用,如TGF-β,當(dāng)組織遭受損傷出現(xiàn)炎癥反應(yīng),TGF-β大量表達,能夠抑制炎癥反應(yīng),維持免疫穩(wěn)態(tài),避免造成組織損傷;但持續(xù)過度表達又造成細(xì)胞外基質(zhì)合成增加,組織纖維化[59,60]。因此,在重建腎臟“防御體系”時必須把握好治療的時機和強度,既能減輕組織炎癥反應(yīng),縮短炎癥持續(xù)時間,同時又不減弱炎癥的保護性作用;既能促進受損組織恢復(fù),又不致引起組織修復(fù)過度。
三、血管活性物質(zhì)
當(dāng)各種因素造成血管內(nèi)皮損傷時,血管內(nèi)皮細(xì)胞合成釋放或活化多種血管活性物質(zhì),調(diào)節(jié)血管舒縮性,活化血小板,促進單核細(xì)胞黏附,血栓生成,脂質(zhì)代謝紊亂,炎癥反應(yīng)發(fā)生,血管重塑等一系列病理生理改變,參與腎臟疾病的發(fā)生和發(fā)展。
目前已知的血管活性肽有血管緊張素、腎上腺素、去甲腎上腺素、內(nèi)皮舒張因子(如NO)、內(nèi)皮素、血管升壓素、心房鈉尿肽、激肽、組胺、前列腺素、阿片肽等,它們大多來源于大分子前體肽原。不同的血管活性肽分子和同一肽原體內(nèi)的眾多酶解片段共同構(gòu)成了極其復(fù)雜的調(diào)節(jié)網(wǎng)絡(luò)。血管內(nèi)皮是內(nèi)皮素、腎上腺素、NO等血管活性物質(zhì)的主要來源;血管外膜成纖維細(xì)胞可分泌干細(xì)胞生長因子、NO等;單核/巨噬細(xì)胞除分泌細(xì)胞因子外,還分泌生長因子、血管活性肽等,調(diào)節(jié)自身和周圍細(xì)胞的功能。
1.腎素-血管緊張素-醛固酮系統(tǒng)
腎素-血管緊張素-醛固酮系統(tǒng)(RAAS)在慢性腎臟病和心血管系統(tǒng)疾病的發(fā)生和進展中發(fā)揮重要作用。血管緊張素Ⅱ(AngⅡ)是RAAS系統(tǒng)最主要的效應(yīng)分子,通過與細(xì)胞膜上的1型受體(AT1)結(jié)合而發(fā)揮作用。AngⅡ不僅可以改變腎小球血流動力學(xué)促進腎小球硬化,還促進系膜細(xì)胞、內(nèi)皮細(xì)胞和腎小管上皮細(xì)胞增生、TGF-β表達及細(xì)胞外基質(zhì)積聚。近年來研究還發(fā)現(xiàn),AngⅡ可以誘導(dǎo)巨噬細(xì)胞炎癥因子IL-6、IL-1β和TNF-α的生成,導(dǎo)致炎癥應(yīng)答產(chǎn)生C-反應(yīng)蛋白[61]。臨床研究發(fā)現(xiàn),血管緊張素Ⅱ受體拮抗劑(ARBs)具有獨立于降壓之外的腎保護作用。有學(xué)者提出,糖尿病患者即使血壓正常也應(yīng)盡早應(yīng)用ARB類藥物以期保護腎功能。AT1受體拮抗劑氯沙坦治療早期糖尿病腎病其機制之一可能就是通過減少ICAM-1的產(chǎn)生,抑制炎癥反應(yīng)而保護腎臟。動物體內(nèi)實驗發(fā)現(xiàn),早期用纈沙坦對糖尿病大鼠進行干預(yù),糖尿病大鼠尿蛋白排泄明顯減少,肌酐清除率增加,腎臟病理學(xué)改變減輕,腎組織巨噬細(xì)胞浸潤減少,提示纈沙坦的腎保護功能可能與改善炎癥微環(huán)境有關(guān)[62]。新近研究表明,RAAS中所有基因都受到Wnt/β-catenin信號通路所調(diào)控。在多柔比星誘導(dǎo)的小鼠腎病模型中發(fā)現(xiàn),應(yīng)用β-catenin的小分子抑制劑ICG-001能夠有效降低RAAS的活化,抑制炎癥反應(yīng)和腎臟纖維化進程[63]。
2.內(nèi)皮素
內(nèi)皮素(ET)最初是由日本學(xué)者Yanagisawa等于1988年在培養(yǎng)的豬主動脈內(nèi)皮細(xì)胞中分離提純的,是目前所知機體內(nèi)最強的縮血管活性多肽。ET通過識別其特異的受體,內(nèi)皮素受體-A(ET-A受體)或內(nèi)皮素受體-B(ET-B受體)而調(diào)節(jié)血管收縮或舒張。內(nèi)皮素至少有三種異構(gòu)體,ET1、ET2和ET3,其中ET-1的生理功能最強[64,65]。
腎臟組織多種細(xì)胞可以合成分泌ET1,如血管平滑肌細(xì)胞及內(nèi)皮細(xì)胞、腎小球系膜細(xì)胞、腎小球臟層上皮細(xì)胞、腎小管上皮細(xì)胞、集合管上皮細(xì)胞、成纖維細(xì)胞、巨噬細(xì)胞等。這些細(xì)胞在分泌ET1的同時又表達ET1受體。ET1通過自分泌和旁分泌作用發(fā)揮多種生理病理學(xué)效應(yīng)。ET1能使腎臟血管產(chǎn)生強大收縮作用,減少腎血流量,降低腎小球濾過率;促使系膜細(xì)胞收縮減少腎小球濾過面積和超濾系數(shù);ET-1還能刺激腎小管上皮細(xì)胞自分泌ET-1并促進其增殖。在胸腺細(xì)胞抗體誘發(fā)的大鼠系膜增生性腎炎中,腎小球產(chǎn)生的ET1明顯增加。抑制ET-1基因的表達,能夠抑制大鼠系膜細(xì)胞和系膜基質(zhì)增生。提示ET1是刺激腎臟組織系膜細(xì)胞和系膜基質(zhì)增生的重要炎癥介質(zhì)之一。此外,ET1還能促進腎間質(zhì)成纖維細(xì)胞增生,上調(diào)Ⅰ型膠原、TGF-β、組織金屬蛋白酶抑制物-1(TIMP-1)的表達,促進血管纖維化;同時趨化單核細(xì)胞并刺激其產(chǎn)生炎癥介質(zhì),如上調(diào)ICAM-1的表達,促進MCP-1的釋放等[54]。
四、黏附分子
近來免疫病理學(xué)研究已經(jīng)證實黏附分子為炎癥過程的樞紐,腎臟疾病與其關(guān)系密切。黏附分子由細(xì)胞產(chǎn)生,存在于細(xì)胞表面,介導(dǎo)細(xì)胞與細(xì)胞間或細(xì)胞與基質(zhì)間相互接觸和結(jié)合。根據(jù)結(jié)構(gòu)和功能不同,黏附分子主要分為四個家族,即免疫球蛋白家族、選擇素家族、整合素家族和鈣黏蛋白家族。
1.免疫球蛋白超家族(immunoglobulin superfamily)
在介導(dǎo)細(xì)胞相互識別、相互作用的黏附分子中,有許多分子具有與免疫球蛋白V區(qū)或C區(qū)相似的折疊結(jié)構(gòu),其氨基酸組成也有一定的同源性,將這類分子歸為免疫球蛋白超家族。其中以ICAM和VCAM為代表。ICAM包括ICAM-1、ICAM-2和ICAM-3,在體內(nèi)分布差異較大。ICAM-1分布最廣,可在多種細(xì)胞上表達,如巨噬細(xì)胞、血管內(nèi)皮細(xì)胞、腎小球上皮細(xì)胞、成纖維細(xì)胞等。VCAM則主要在活化的血管內(nèi)皮細(xì)胞表達。IL-1、TNF-α、IFN、脂多糖等炎癥信號均可上調(diào)上述兩類分子的表達。炎癥時,活化的內(nèi)皮細(xì)胞表面的ICAM、VCAM可與中性粒細(xì)胞、巨噬細(xì)胞表面相應(yīng)的受體結(jié)合,繼選擇素介導(dǎo)的中性粒細(xì)胞、巨噬細(xì)胞與內(nèi)皮細(xì)胞的黏附作用之后,使其固著于炎癥部位的血管內(nèi)皮細(xì)胞上,分泌水解酶,破壞內(nèi)皮細(xì)胞,從血管中穿出游走入組織。
在增殖性腎炎和IgA腎病的腎活檢標(biāo)本中,ICAM-1的表達彌散分布,與腎組織內(nèi)浸潤的單核細(xì)胞和巨噬細(xì)胞的數(shù)量明顯相關(guān)。在病理改變較重的狼瘡性腎炎和紫癜性腎炎的腎臟活檢標(biāo)本中也可見ICAM-1的高表達以及T淋巴細(xì)胞和巨噬細(xì)胞的浸潤[66]。在糖尿病腎病、單側(cè)輸尿管梗阻、中毒性腎炎、缺血-再灌注損傷的動物模型中也發(fā)現(xiàn)腎組織內(nèi)ICAM-1表達明顯上調(diào),而將ICAM-1基因敲除后病變則明顯減輕[67]。因此ICAM-1可看作是腎臟炎癥活動的主要標(biāo)志之一。
CD146又名黑色素瘤黏附分子,也是免疫球蛋白超家族的成員之一。CD146在正常腎組織中主要表達于內(nèi)皮細(xì)胞、平滑肌細(xì)胞、系膜細(xì)胞。在慢性腎衰竭患者局部腎組織中,CD146不僅在上述組織中高表達,同時還在腎小管上皮細(xì)胞表達。進一步研究發(fā)現(xiàn),CD146參與了白細(xì)胞的活化,能夠促進炎癥細(xì)胞向內(nèi)皮細(xì)胞黏附、貼壁、游走,從而參與腎間質(zhì)纖維化過程[68]。
2.選擇素家族(selectin family)
目前已發(fā)現(xiàn)選擇素家族中有三個成員:L-選擇素、P-選擇素和E-選擇素,其中對P-選擇素研究較多。P-選擇素位于血小板的α-顆粒和血管內(nèi)皮細(xì)胞的Weibel-Palade小體內(nèi),它的配體表達于所有中性粒細(xì)胞、單核細(xì)胞、巨噬細(xì)胞、淋巴細(xì)胞等表面。靜息狀態(tài)下,血小板和內(nèi)皮細(xì)胞表面不表達P-選擇素,一旦被TGF-β、高糖等激活后,α-顆粒和Weibel-Palade小體迅速與細(xì)胞膜融合,使P-選擇素在血小板和血管內(nèi)皮細(xì)胞表面表達,介導(dǎo)活化的血小板、內(nèi)皮細(xì)胞、粒細(xì)胞、單核細(xì)胞的黏附,啟動一系列炎癥反應(yīng)。
近年來對選擇素在腎臟病中作用的研究結(jié)果并不一致。Hirata等報道在糖尿病腎病模型中,腎小球內(nèi)P-選擇素表達上調(diào)與炎性細(xì)胞浸潤數(shù)量顯著相關(guān)。人工合成的選擇素阻斷劑SKK60037可顯著抑制大鼠血栓性腎炎模型中的腎小球病變。然而Rosenkranz等利用P-選擇素基因打靶鼠構(gòu)建抗GBM腎炎模型,發(fā)現(xiàn)尿蛋白含量及腎小球病變并未減輕反而加重,說明選擇素除了介導(dǎo)炎性細(xì)胞黏附外尚具有其他的重要功能,因此選擇素在不同的腎臟疾病及其不同的發(fā)展階段中所起的作用可能是不同的[54]。
3.整合素家族(integrin family)
整合素家族是一組介導(dǎo)細(xì)胞黏附的細(xì)胞表面糖蛋白受體,其主要功能為介導(dǎo)細(xì)胞黏附和信號轉(zhuǎn)導(dǎo),參與腎臟發(fā)育、整體結(jié)構(gòu)維持、細(xì)胞增生和基質(zhì)更新以及新陳代謝等調(diào)節(jié),其異常表達及其黏附機制紊亂在腎臟疾病中具有重要意義。腎間質(zhì)纖維化是各種原因造成腎小管及間質(zhì)病變的最終結(jié)果,也是導(dǎo)致終末期腎衰竭原因之一,該進程的主要病理特征是間質(zhì)成纖維細(xì)胞增殖和ECM的過度積聚。整合素通過細(xì)胞黏附及其信號轉(zhuǎn)導(dǎo)機制,參與小管上皮細(xì)胞EMT,激活成纖維細(xì)胞轉(zhuǎn)化成肌纖維細(xì)胞并增殖,而致間質(zhì)纖維化。研究表明腎損害時整合素介導(dǎo)炎癥細(xì)胞尤其巨噬細(xì)胞與內(nèi)皮細(xì)胞或ECM黏附而使其激活,釋放大量的IL-1、TGF-β、結(jié)締組織生長因子(CTGF)等細(xì)胞因子和生長因子,促進腎間質(zhì)纖維化[69]。
4.鈣黏蛋白家族(cadherin family)
鈣黏蛋白是一類Ⅰ型跨膜蛋白,是細(xì)胞連接、細(xì)胞黏附必不可少的分子,因其功能發(fā)揮需要鈣離子,由此而得名。目前已知鈣黏蛋白家族共有三個成員,E-鈣黏蛋白,N-鈣黏蛋白和P-鈣黏蛋白。不同的鈣黏蛋白分子在體內(nèi)分布存在差異,且表達水平隨細(xì)胞的分化、發(fā)育狀態(tài)不同而改變。E-鈣黏蛋白、N-鈣黏蛋白表達于腎小管上皮細(xì)胞,對維持腎小管上皮細(xì)胞極性和完整性非常重要;N-鈣黏蛋白也在系膜細(xì)胞中大量表達,通過與α-catenin、β-catenin和肌動蛋白相連,以維持系膜細(xì)胞之間以及與球旁細(xì)胞之間的縫隙連接,對穩(wěn)定毛細(xì)血管袢的內(nèi)壓和濾過率非常重要。當(dāng)腎臟組織遭受損傷時,鈣黏蛋白會有不同程度的丟失。如在急性腎損傷患者和缺血-再灌注大鼠模型中,均發(fā)現(xiàn)近端腎小管N-鈣黏蛋白表達明顯下調(diào)[70]。慢性腎病患者體內(nèi),長期的TGF-β刺激,也導(dǎo)致腎小管上皮細(xì)胞E-鈣黏蛋白表達減少,細(xì)胞完整性遭到破壞。有研究發(fā)現(xiàn)糖尿病腎病患者的鈣黏蛋白排泄量隨著腎病的加重明顯增加,推測E-鈣黏蛋白有可能成為一個新的糖尿病腎病的診斷標(biāo)志物[71]。
五、炎癥小體
炎癥小體(in flammasome)是由NOD樣受體家族(NLRs)參與組裝的多蛋白水解復(fù)合物,此概念由Tschopp研究小組于2002年首次提出[72]。炎癥小體能夠識別病原相關(guān)分子模式(pathogenassociated molecular patterns,PAMPs)或者宿主來源的危險信號分子(damage/danger-associated molecular patterns,DAMPs),通過誘導(dǎo)細(xì)胞活化、炎癥介質(zhì)釋放,引起炎癥反應(yīng)(圖2-4-2-2)。由于能被多種類型的病原體或危險信號所激活,因此炎癥小體參與眾多疾病的發(fā)生發(fā)展過程。
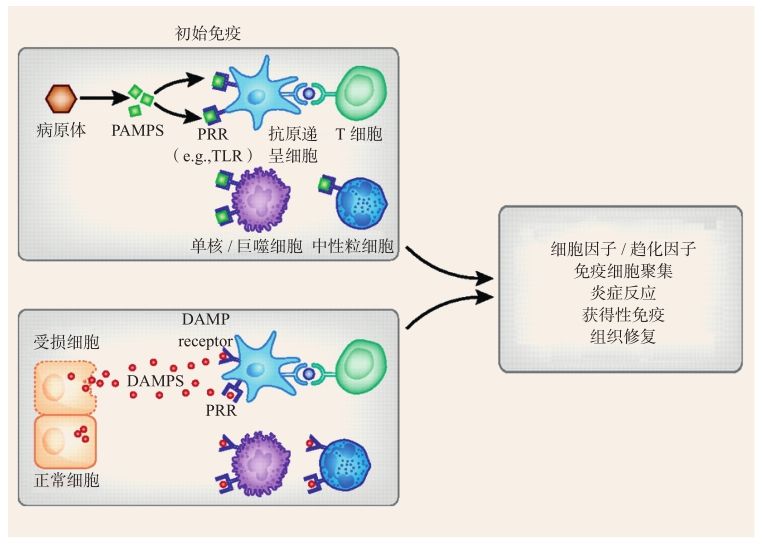
圖2-4-2-2 炎癥小體介導(dǎo)炎癥反應(yīng)發(fā)生[73]
目前已有大量證據(jù)表明炎癥小體參與多種類型的腎臟疾病,并在多個階段發(fā)揮作用。比如缺血引起的急性腎損傷,近端小管受損產(chǎn)生DAMPs,識別炎癥小體中的Toll樣受體(TLRs)和晚期糖化終產(chǎn)物受體(receptor for advanced glycation end products,RAGE),高遷移率族蛋白1(highmobility group box 1,HMGB1)、熱休克蛋白(HSPs),促進促炎因子、趨化因子的釋放,誘導(dǎo)抗原遞呈細(xì)胞的成熟和遷移;同時HSPs還通過增強MHCⅠ類分子的表達而促進T細(xì)胞的活化[73]。細(xì)胞外基質(zhì)中的蛋白聚糖是DAMP中一個促炎因子,給小鼠單側(cè)輸尿管結(jié)扎后4天,即可在小管上皮細(xì)胞中檢測到二聚糖的升高,同時NLRP3炎癥小體被激活,刺激體內(nèi)產(chǎn)生促炎因子IL-1β;而在二聚糖缺失的小鼠體內(nèi),NLRP3炎癥小體和IL-1β則被抑制[74]。
臨床檢測發(fā)現(xiàn),在IgA腎病、微小病變腎病、膜性腎病、局灶節(jié)段性腎小球硬化癥、狼瘡性腎炎、高血壓腎病、新月體腎炎及急性腎小管壞死,均有NLRP3表達顯著上調(diào),且表達量與患者血肌酐水平呈正相關(guān),提示炎癥小體參與急、慢性腎臟疾病[75]。
第三節(jié) 炎癥介導(dǎo)的腎臟進行性損傷的病理過程
腎臟疾病,大體可分為急性腎損傷(AKI)和慢性腎臟病(CKD)。臨床上,部分急性腎損傷在經(jīng)過治療后可達到臨床痊愈或恢復(fù)正常,但相當(dāng)比例的急性腎損傷會逐漸發(fā)展成為慢性腎臟病,最終進展為終末期腎病(ESRD)。無論是急性腎損傷還是慢性腎臟病,或兩者之間的演變,在腎臟進行性損傷的病理過程中,炎癥在各個環(huán)節(jié)發(fā)揮著重要作用;它既是引起腎臟損傷的始動因素,又是促進和加重腎臟損傷的推動力量。在本節(jié),我們將重點討論炎癥細(xì)胞浸潤與腎小管間質(zhì)損傷之間的關(guān)系。首先需要明確的是,進展性腎間質(zhì)纖維化典型的病理學(xué)特征是大量的炎性細(xì)胞浸潤、間質(zhì)腎小管萎縮、肌成纖維細(xì)胞激活及其導(dǎo)致的細(xì)胞外基質(zhì)過度堆積,最終取代正常的腎臟結(jié)構(gòu),造成腎臟功能的永久性喪失或衰竭[76,77]。這一系列的病理變化,主要由聚集于腎小管間質(zhì)的炎性細(xì)胞及其分泌的炎癥因子所介導(dǎo)。具體來說,可以將進展性腎臟病理損傷分為以下四個時期(圖2-4-3-1):
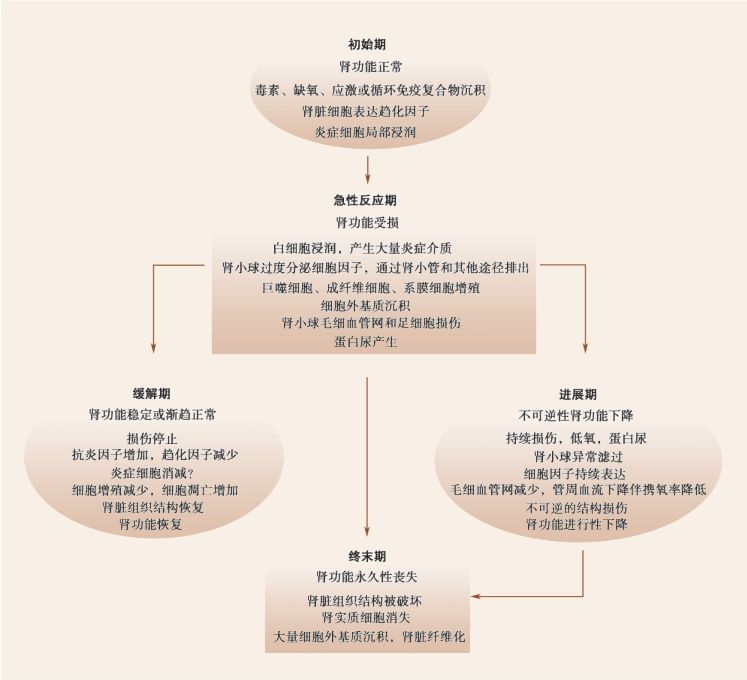
圖2-4-3-1 炎癥反應(yīng)介導(dǎo)腎臟疾病的發(fā)生發(fā)展
一、初始期
腎臟組織固有細(xì)胞一旦遭受外界刺激,無論是物理、化學(xué)或生物損傷,首要反應(yīng)是產(chǎn)生促炎分子,募集炎癥細(xì)胞聚集于受損部位,誘發(fā)炎癥反應(yīng)。由于組織表達特定的黏附分子、趨化因子,因此募集來炎癥細(xì)胞以初級免疫反應(yīng)細(xì)胞為主,如中性粒細(xì)胞、單核/巨噬細(xì)胞等;且此時的炎癥反應(yīng)程度較弱,范圍局限。
二、急性反應(yīng)期
在急性反應(yīng)期,腎臟受損部位所浸潤的炎癥細(xì)胞可進一步釋放細(xì)胞因子、趨化因子等,活化T細(xì)胞等次級免疫反應(yīng)細(xì)胞,進一步擴大炎癥反應(yīng);同時造成局部損傷加重。諸如,在腎小管間質(zhì)疾病中,活化的巨噬細(xì)胞一方面可直接促進細(xì)胞外基質(zhì)的形成和聚集,另一方面其還通過分泌一系列生長因子,如成纖維細(xì)胞生長因子(FGF)、TGF-β、TNF-α、表皮生長因子(EGF)和PDGF等,造成成纖維細(xì)胞的異常增殖,并最終導(dǎo)致間質(zhì)區(qū)域擴大和間質(zhì)纖維化[78]。此階段的成纖維細(xì)胞主要有三個來源,其中大部分是來自于間質(zhì)中原有的成纖維細(xì)胞和血管周細(xì)胞(pericyte)的激活;部分來自于循環(huán)血液中尚未分化成熟的單核樣纖維細(xì)胞( fibrocyte),當(dāng)腎臟局部受損時,這部分細(xì)胞從血管中游走出來,聚集于損傷部位,逐漸分化為成熟的成纖維細(xì)胞[79];還有部分成纖維細(xì)胞來自于經(jīng)歷了EMT的腎小管上皮細(xì)胞和血管內(nèi)皮細(xì)胞[80]。
而在腎小球疾病中,由活化的巨噬細(xì)胞所分泌產(chǎn)生的細(xì)胞因子還可導(dǎo)致腎小球系膜細(xì)胞增生,細(xì)胞外基質(zhì)合成增加,引起典型的系膜增生性腎小球腎炎;炎癥細(xì)胞分泌的細(xì)胞因子還可導(dǎo)致足細(xì)胞結(jié)構(gòu)發(fā)生改變,足突廣泛融合,引發(fā)蛋白尿產(chǎn)生;融合的足突與腎小球基底膜粘連,形成局灶節(jié)段性腎小球硬化。此外,在原發(fā)性腎小球腎炎,如膜性腎小球腎炎、局灶節(jié)段性腎小球硬化、系膜增生性腎小球腎炎中均可觀察到腎間質(zhì)中有大量的成纖維細(xì)胞增殖。那么,腎小球疾病如何引起腎間質(zhì)成纖維細(xì)胞的活化和增殖呢?在這一過程中,腎小管上皮細(xì)胞發(fā)揮著極其重要的調(diào)節(jié)作用。腎小球受損后,大量分泌的炎癥因子、生長因子或者產(chǎn)生的尿白蛋白,在通過腎小管排出時可激惹腎小管細(xì)胞進一步分泌促炎、促纖維化的生長因子、細(xì)胞因子和趨化因子等,并通過腎小球-腎小管周圍循環(huán)毛細(xì)血管網(wǎng)進一步促進了間質(zhì)炎癥細(xì)胞的浸潤和聚集,從而加速了腎小球疾病中腎小管間質(zhì)纖維化的進程[81]。
三、緩解期或進展期
如果機體有良好的免疫調(diào)節(jié)功能,那么腎臟組織在經(jīng)歷了急性炎癥反應(yīng),及時消除了病原且不再繼續(xù)遭受損傷,炎癥反應(yīng)會逐漸消退,此時有賴于一系列抑制炎癥反應(yīng)的炎癥細(xì)胞和炎癥介質(zhì)。如巨噬細(xì)胞會發(fā)生表型轉(zhuǎn)換,從促炎的M1型轉(zhuǎn)換為抗炎的M2型,分泌TGF-β、IL-10等抑制炎癥反應(yīng)。在TGF-β、IL-4等抗炎細(xì)胞因子的作用下,原始CD4+T細(xì)胞向促炎型的Th1細(xì)胞分化減少,轉(zhuǎn)而向Th2、抑制性Treg細(xì)胞分化增多,這些細(xì)胞進一步分泌TGF-β、IL-4等抑制炎癥反應(yīng)。此外,組織細(xì)胞受損減弱,表達的黏附分子、趨化因子也逐步減少,炎癥細(xì)胞不再大量聚集,逐漸從活化狀態(tài)恢復(fù)為靜息狀態(tài)。由此,炎癥反應(yīng)逐漸消退。組織受損輕微的細(xì)胞開始修復(fù),受損嚴(yán)重細(xì)胞走向凋亡,被新增殖的細(xì)胞所取代。腎臟組織結(jié)構(gòu)和腎臟功能逐步恢復(fù)。
然而,如果外來損傷持續(xù)加重,或炎癥反應(yīng)未能及時得到有效控制,那么,腎臟固有細(xì)胞則產(chǎn)生大量細(xì)胞外基質(zhì),最終導(dǎo)致腎臟出現(xiàn)不可逆的結(jié)構(gòu)和功能的改變。此時的腎臟組織的炎癥狀態(tài)不同于急性反應(yīng)期的劇烈,而是一種相對較弱且持久不衰的狀態(tài),促炎、抗炎細(xì)胞共存且以抗炎細(xì)胞為主,抗炎細(xì)胞分泌大量促纖維化因子,促進組織纖維化。在急性腎損傷中,由于腎小管間質(zhì)缺血可誘導(dǎo)腎小管細(xì)胞凋亡、壞死和炎癥細(xì)胞浸潤,當(dāng)疾病轉(zhuǎn)歸進入慢性期后,腎小管間質(zhì)中浸潤的巨噬細(xì)胞、T細(xì)胞持續(xù)分泌促纖維化因子,可誘導(dǎo)成纖維細(xì)胞進一步擴增,產(chǎn)生大量細(xì)胞外基質(zhì)[82,83]。而活化的上皮細(xì)胞也同時分泌產(chǎn)生細(xì)胞外基質(zhì)和細(xì)胞因子、趨化因子等,在損傷過程中,部分上皮細(xì)胞可轉(zhuǎn)分化為肌成纖維細(xì)胞并遷移到間質(zhì)區(qū)域。間質(zhì)細(xì)胞的自身浸潤以及大量細(xì)胞外基質(zhì)的沉積,導(dǎo)致間質(zhì)區(qū)域明顯擴大,使得腎小管周圍毛細(xì)血管遠(yuǎn)離腎小管,破壞了氧氣的彌散、腎小管重吸收和分泌功能。在腎小球內(nèi),浸潤的巨噬細(xì)胞可刺激系膜細(xì)胞分泌Ⅳ型膠原、層粘連蛋白、纖維連接蛋白;引起系膜細(xì)胞擴張;擴張的系膜細(xì)胞導(dǎo)致腎小球毛細(xì)血管叢狹窄或閉塞,逐漸導(dǎo)致足細(xì)胞損傷,腎小球硬化以及腎小管周圍毛細(xì)血管的破壞,腎小管間質(zhì)結(jié)構(gòu)異常[84]。
四、終末期
長期的慢性炎癥刺激,導(dǎo)致腎臟毛細(xì)血管數(shù)目明顯減少,組織彌漫性瘢痕形成,腎小管萎縮,腎小球硬化。腎實質(zhì)細(xì)胞顯著減少,細(xì)胞外基質(zhì)大量沉積,腎臟正常組織結(jié)構(gòu)消失,功能永久性喪失,臨床進入終末期腎病階段。
綜上,由于腎臟局部損傷,表達和釋放炎癥介質(zhì),誘導(dǎo)炎癥細(xì)胞浸潤,啟動局部炎癥反應(yīng)。如果此時炎癥僅局限在一定范圍內(nèi)并能夠及時消退,則疾病經(jīng)歷了急性期后逐漸轉(zhuǎn)歸正常;但如果炎癥不能得到有效控制,或組織再次遭受損傷,則炎癥反應(yīng)不斷加重和擴大,造成周圍組織損傷,同時誘導(dǎo)腎臟固有細(xì)胞也積極參與炎癥反應(yīng),促使整個腎臟成為一個炎癥體,導(dǎo)致腎臟固有細(xì)胞丟失,成纖維細(xì)胞擴增,細(xì)胞外基質(zhì)沉積,腎臟正常結(jié)構(gòu)、功能被破壞,逐步走向纖維化。
結(jié)束語
目前對大多數(shù)腎臟疾病的發(fā)病機制和發(fā)展過程的認(rèn)識還很有限,但毫無疑問炎癥細(xì)胞和炎癥介質(zhì)介導(dǎo)的調(diào)控網(wǎng)絡(luò)在腎臟疾病的發(fā)生、發(fā)展、轉(zhuǎn)歸和預(yù)后過程中都發(fā)揮著重要作用。深入認(rèn)識和闡明其中的具體機制,尋找關(guān)鍵的始動因素和重要的節(jié)點事件,進行特定的干預(yù)和治療,均衡促炎、抗炎兩大勢力的強弱,控制炎癥反應(yīng)的強度,促進炎癥反應(yīng)及時消退,將成為未來炎癥相關(guān)性腎臟疾病治療的有效策略。
(傅海燕 劉友華)
參考文獻
1.KINSEY GR, LI L, OKUSA MD. Inflammation in Acute Kidney Injury. Nephron Exp Nephrol, 2008,109(4):e102-107.
2.MENG XM, NIKOLIC-PATERSON DJ, LAN HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol,2014, 10(9):493-503.
3.BOLISETTY S, AGARWAL A. Neutrophils in acute kidney injury:not neutral any more. Kidney Int, 2009,75(7):674-676.
4.WYNN TA, CHAWLA A, POLLARD JW. Macrophage biology in development, homeostasis and disease.Nature, 2013, 496(7446):445-455.
5.WANG Y, HARRIS DC. Macrophages in renal disease. J Am Soc Nephrol, 2011, 22(1):21-27.
6.SASSY-PRIGENT C, HEUDES D, MANDET C, et al. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes, 2000, 49(3):466-475.
7.KATO S, LUYCKX VA, OTS M, et al. Renin-angiotensin blockade lowers MCP-1 expression in diabetic rats.Kidney Int, 1999, 56(3):1037-1048.
8.CHOW F, OZOLS E, NIKOLIC-PATERSON DJ, et al. Macrophages in mouse type 2 diabetic nephropathy:correlation with diabetic state and progressive renal injury. Kidney Int, 2004, 65(1):116-128.
9.GORDON S, TAYLOR PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol, 2005, 5(12):953-964.
10.KLUTH DC. Pro-resolution properties of macrophages in renal injury. Kidney Int, 2007, 72(3):234-236.
11.GORDON S. Alternative activation of macrophages. Nat Rev Immunol, 2003, 3(1):23-35.
12.BOURLIER V, ZAKAROFF-GIRARD A, MIRANVILLE A, et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation, 2008, 117(6):806-815.
13.RICARDO SD, VAN GOOR H, EDDY AA. Macrophage diversity in renal injury and repair. J Clin Invest,2008, 118(11):3522-3530.
14.DUFFIELD JS, TIPPING PG, KIPARI T, et al. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol, 2005, 167(5):1207-1219.
15.WANG Y, WANG YP, ZHENG G, et al. Ex vivo programmed macrophages ameliorate experimental chronic in flammatory renal disease. Kidney Int, 2007, 72(3):290-299.
16.CAO YL, WANG YX, WANG DF, et al. Correlation between omental TNF-α protein and plasma PAI-1 in obesity subjects. Int J Cardiol, 2008, 128(3):399-405.
17.MATOBA K, KAWANAMI D, ISHIZAWA S, et al. Rho-kinase mediates TNF-α-induced MCP-1 expression via p38 MAPK signaling pathway in mesangial cells. Biochem Biophys Res Commun, 2010, 402(4):725-730.
18.LEE DL, LEITE R, FLEMING C, et al. Hypertensive response to acute stress is attenuated in interleukin-6 knockout mice. Hypertension, 2004, 44(3):259-263.
19.PAWARIA S, RAMANI K, MAERS K, et al. Complement component C5a permits the coexistence of pathogenic Th17 cells and type I IFN in Lupus. J Immunol, 2014, 193(7): 3288-3295.
20.林芙君,劉秀君,張文竹,等.白細(xì)胞介素17在IgA腎病腎小管間質(zhì)的表達及意義.臨床腎臟病雜志,2010,10(10):453-456.
21.MATSUMOTO K, KANMATSUSE K. Interleukin-17 stimulates the release of pro-in flammatory cytokines by blood monocytes in patients with IgA nephropathy. Scand J Urol Nephrol, 2003, 37(2):164-171.
22.VIGNALI DA, COLLISON LW, WORKMAN CJ. How regulatory T cells work. Nat Rev Immunol, 2008(7),8:523-532.
23.LE BERRE L, BRUNEAU S, NAULET J, et al. Induction of T regulatory cells attenuates idiopathic nephrotic syndrome. J Am Soc Nephrol, 2009, 20(1):57-67.
24.XING Q, SU H, CUI J, et al. Role of Treg cells and TGF-β1 in patients with systemic lupus erythematosus:a possible relation with lupus nephritis. Immunol Invest, 2012, 41(1):15-27.
25.TURLEY SJ. Dendritic cell: Inciting and inhibiting autoimmunity. Curr Opin Immunol, 2002, 14(6):765-770.
26.吳俊逸,張薇.終末期腎病維持性血透患者樹突狀細(xì)胞分化成熟能力的研究. 上海交通大學(xué)學(xué)報醫(yī)學(xué)版.2013,33(3):290-293.
27.TORZICKY M, VIZNEROVA P, RICHTER S, et al. Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) and CD99 are critical in lymphatic transmigration of human dendritic cells. J Invest Dermatol, 2012,132(4):1149-1157.
28.BASILE DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function.Kidney Int, 2007, 72(2):151-156.
29.KANWAR YS, WADA J, SUN L, et al. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med, 2008, 233(1):4-11.
30.ZIMERING MB, ZHANG JH, GUARINO PD, et al. Endothelial cell autoantibodies in predicting declining renal function, end-stage renal disease, or death in adult type 2 diabetic nephropathy. Front Endocrinol, 2014,5(5):128.
31.PR?LS F, HARTNER A, SCH?CKLMANN HO, et al. Mesangial cells and their adhesive properties. Exp Nephrol, 1999, 7(2):137-146.
32.PICKEN MM. The role of mesangial homeostasis in glomerular injury progression: hope for mesangial sclerosis reversal. Kidney Int, 2009, 75(6):574-576.
33.MIGLIORINI A, EBID R, SCHERBAUM CR, et al. The danger control concept in kidney disease: mesangial cells. J Nephrol, 2013, 26(3):437-449.
34.YUNG S, CHEUNG KF, ZHANG Q, et al. Mediators of in flammation and their effect on resident renal cells:implications in lupus nephritis. Clin Dev Immunol. 2013,(2):317682.
35.BOOR P, OSTENDORF T, FLOEGE J. Renal fibrosis: novel insights into mechanisms and therapeutic targets.Nat Rev Nephrol, 2010, 6(11):643-656.
36.LOPEZ-NOVOA JM, NIETO MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med, 2009, 1:303-314.
37.LI Y, WEN X, LIU Y. Tubular cell dedifferentiation and peritubular inflammation are coupled by the transcription regulator Id1 in renal fibrogenesis. Kidney Int, 2012, 81(9):880-891.
38.BR?HLER S1, ISING C, HAGMANN H, et al. Intrinsic proin flammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria. Am J Physiol Renal Physiol, 2012, 303(10):F1473-1485.
39.MIGLIORINI A1, ANGELOTTI ML, MULAY SR, et al. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am J Pathol, 2013, 183(2):431-440.
40.SCHIFFER M, BITZER M, ROBERTS IS, et al. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest, 2001, 108(6):807-816.
41.李金紅,陶建瓴,李航.足細(xì)胞損傷與糖尿病腎病的研究現(xiàn)狀. 中國醫(yī)學(xué)科學(xué)院學(xué)報. 2010,32(5):590-596.
42.CHUNG AC, LAN HY. Chemokines in renal injury. J Am Soc Nephrol, 2011, 22(5):802-809.
43.SCHL?NDORFF D, NELSON PJ, LUCKOW B, et al. Chemokines and renal disease. Kidney Int, 1997,51(3):610-621.
44.LI Y, TUCCI M, NARAIN S, et al. Urinary biomarkers in lupus nephritis. Autoimmun Rev, 2006, 5(6):383-388.
45.PANZER U, STEINMETZ OM, STAHL RA, et al. Kidney diseases and chemokines. Curr Drug Targets, 2006,7(1):65-80.
46.VIANNA HR, SOARES CM, SILVEIRA KD, et al. Cytokines in chronic kidney disease: potential link of MCP-1 and dyslipidemia in glomerular diseases. Pediatr Nephrol, 2013, 28(3):463-469.
47.BADAL SS, DANESH FR. New insights into molecular mechanisms of diabetic kidney disease. Am J Kidney Dis, 2014, 63(2): 63-83.
48.RAMESH G, REEVES WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest, 2002, 110(6):835-842.
49.KNOTEK M, ROGACHEV B, WANG W, et al. Endotoxemic renal failure in mice: Role of tumor necrosis factor independent of inducible nitric oxide synthase. Kidney Int, 2001, 59(6):2243-2249.
50.KIR HM, ERALDEMIR C, DERVISOGLU E, et al. Effects of chronic kidney disease and type of dialysis on serum levels of adiponectin, TNF-alpha and high sensitive C-reactive protein. Clin Lab, 2012, 58:495-500.
51.LICHTNEKERT J, KULKARNI OP, MULAY SR, et al. Anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3/ASC in flammasome-mediated activation of caspase-1. PLoS One, 2011, 6(10):e26778.
52.KARKAR AM, TAM FW, STEINKASSERER A, et al. Modulation of antibody-mediated glomerular injury in vivo by IL-1ra, soluble IL-1 receptor, and soluble TNF receptor. Kidney Int, 1995, 48(6):1738-1746.
53.王海燕.腎臟病學(xué). 3版. 北京:人民衛(wèi)生出版社,2008:750-751.
54.ZUBAIR A, FRIERI M. Lupus nephritis: review of the literature. Curr Allergy Asthma Rep, 2013, 13(6):580-586.
55.LIU Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol, 2004, 287(1):F7-16.
56.LAN HY. Diverse roles of TGF-β/Smads in renal fibrosis and in flammation. Int J Biol Sci, 2011, 7(7):1056-1067.
57.GIANNOPOULOU M1, DAI C, TAN X, et al. Hepatocyte growth factor exerts its anti-in flammatory action by disrupting nuclear factor-kappa B signaling. Am J Pathol, 2008, 173(1):30-41.
58.SINUANI I, BEBERASHVILI I, AVERBUKH Z, et al. Role of IL-10 in the progression of kidney disease.World J Transplant, 2013, 3(4):91-98.
59.LIU Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int, 2006, 69(2):213-217.
60.GARCíA-SáNCHEZ O, LóPEZ-HERNáNDEZ FJ, LóPEZ-NOVOA JM. An integrative view on the role of TGF-beta in the progressive tubular deletion associated with chronic kidney disease. Kidney Int, 2010,77(11):950-955.
61.GUO F, CHEN XL, WANG F, et al. Role of angiotensin II type 1 receptor in angiotensin II-induced cytokine production in macrophages. J Interferon Cytokine Res, 2011, 31(4):351-361.
62.唐平,劉丹,楊川,等.纈沙坦對早期糖尿病鼠腎臟巨噬細(xì)胞浸潤的影響. 中華全科醫(yī)學(xué). 2010, 8(2):136-137.
63.ZHOU L, LI Y, HAO S, et al. Multiple genes of the renin-angiotensin system are novel targets of Wnt/β-catenin signaling. J Am Soc Nephrol, 2015, 26(1):107-120.
64.DHAUN N, GODDARD J, WEBB DJ. The endothelin system and its antagonism in chronic kidney disease. J Am Soc Nephrol, 2006, 17(4):943-955.
65.TAN RJ, ZHOU L, ZHOU D, et al. Endothelin receptor A blockade is an ineffective treatment for adriamycin nephropathy. PLoS One, 2013, 8(11):e79963.
66.KUROIWA T, LEE EG. Cellular interactions in the pathogenesis of lupus nephritis: the role of T cells and macrophages in the amplification of the in flammatory process in the kidney. Lupus, 1998, 7(9):597-603.
67.TRONCOSO P, ORTIZ AM, DOMíNGUEZ J, et al. Use of FTY 720 and ICAM-1 antisense oligonucleotides for attenuating chronic renal damage secondary to ischemia-reperfusion injury. Transplant Proc, 2005,37(10):4284-4288.
68.DANIEL L, BARDIN N, MOAL V, et al. Tubular CD146 expression in nephropathies is related to chronic renal failure. Nephron Exp Nephrol, 2005, 99(4):e105-111.
69.孫桂芝,周同.整合素及其與腎臟疾病. 中國中西醫(yī)結(jié)合腎病雜志. 2003,4(6):362-364.
70.NüRNBERGER J, FELDKAMP T, KAVAPURACKAL R, et al. N-cadherin is depleted from proximal tubules in experimental and human acute kidney injury. Histochem Cell Biol, 2010, 133(6):641-649.
71.JIANG H, GUAN G, ZHANG R, et al. Identification of urinary soluble E-cadherin as a novel biomarker for diabetic nephropathy. Diabetes Metab Res Rev, 2009, 25(3):232-241.
72.MARTINON F, BURNS K, TSCHOPP J. The in flammasome: a molecular platform triggering activation of in flammatory caspases and processing of pro-IL-beta. Mol Cell, 2002, 10(2):417-426.
73.ROSIN DL, OKUSA MD. Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol, 2011, 22(3):416-425.
74.BABELOVA A, MORETH K, TSALASTRA-GREUL W, et al. Biglycan, a dangersignal that activates the NLRP3 in flammasomevia Toll and P2X receptors. J Biol Chem, 2009, 284(36): 24035-24048.
75.VILAYSANE A, CHUN J, SEAMONE ME, et al. The NLRP3 Inflammasome Promotes Renal Inflammation and Contributes to CKD. J Am Soc Nephrol, 2010, 21(10):1732-1744.
76.LIU Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol, 2011, 7(12): 684-696.
77.STRUTZ F, MüLLER GA. Interstitial pathomechanisms underlying progressive tubulointerstitial damage.Kidney Blood Press Res, 1999, 22:71-80.
78.TAN RJ, LIU Y. Macrophage-derived TGF-β in renal fibrosis: not a macro-impact after all. Am J Physiol Renal Physiol, 2013, 305(6):F821-822.
79.LEBLEU VS, TADURI G, O’CONNELL J, et al. Origin and function of myo fibroblasts in kidney fibrosis. Nat Med, 2013, 19(8):1047-1053.
80.LIU Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol, 2004, 15(1):1-12.
81.ZHOU D, LI Y, ZHOU L, et al. Sonic hedgehog is a novel tubule-derived growth factor for interstitial fibroblasts after kidney injury. J Am Soc Nephrol. 2014, 25(10):2187-2200.
82.ZHOU D, TAN RJ, LIN L, et al. Activation of hepatocyte growth factor receptor, c-met, in renal tubules is required for renoprotection after acute kidney injury. Kidney Int, 2013, 84(3):509-520.
83.ZHOU D, LI Y, LIN L, et al. Tubule-specific ablation of endogenous β-catenin aggravates acute kidney injury in mice. Kidney Int, 2012, 82(5):537-547.
84.MARCUSSEN N. Tubulointerstitial damage leads to atubularglomeruli: Significance and possible role in progression. Nephrol Dial Transplant, 2000, 15(6):74-75.
- 骨動人生:骨質(zhì)疏松知識讀本
- 鄉(xiāng)村醫(yī)生實用手冊
- 醫(yī)學(xué)影像學(xué)讀片診斷圖譜:胸部分冊
- 感染科護理查房手冊
- 倫理審查體系評估標(biāo)準(zhǔn)
- 中藥材農(nóng)藥殘留研究及國際標(biāo)準(zhǔn)制定
- 高血壓的社區(qū)管理與自我護理
- 醫(yī)院信息系統(tǒng)典型故障案例解析
- 湖北法醫(yī)學(xué)司法鑒定進展
- 常用醫(yī)療專科評估量表
- 中藥化學(xué)實驗方法學(xué)
- 臨床技術(shù)操作規(guī)范:輔助生殖技術(shù)和精子庫分冊(2021修訂版)
- 老年專科護理
- 視力障礙輔助技術(shù)
- 全國縣級醫(yī)院系列實用手冊:感染科醫(yī)生手冊